|
|
Brain Studies
|
A
continuing education course for 30 ces
As the field of psychology understands the importance of neuroscience, www.psychceu.com is pleased to announce a course on the brain, written by leading researchers. Click on the links below to jump to specific sections: Introduction:
Development:
Specific Neurological Conditions:
|
Introduction
Brain
Basics: Know Your Brain
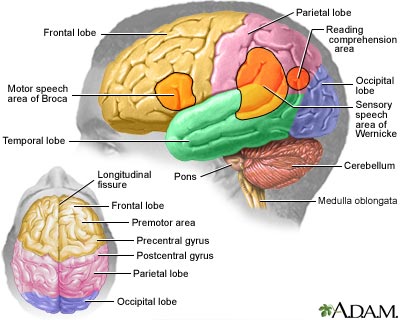
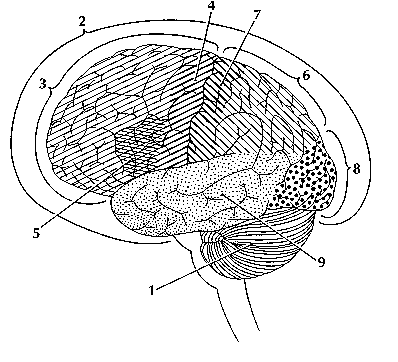
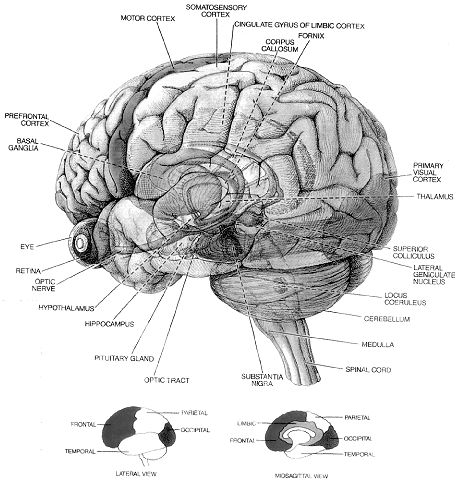
IntroductionThe brain is the most complex part of the human body. This three-pound organ is the seat of intelligence, interpreter of the senses, initiator of body movement, and controller of behavior. Lying in its bony shell and washed by protective fluid, the brain is the source of all the qualities that define our humanity. The brain is the crown jewel of the human body. For centuries, scientists and philosophers have been fascinated by the brain, but until recently they viewed the brain as nearly incomprehensible. Now, however, the brain is beginning to relinquish its secrets. Scientists have learned more about the brain in the last 10 years than in all previous centuries because of the accelerating pace of research in neurological and behavioral science and the development of new research techniques. As a result, Congress named the 1990s the Decade of the Brain. At the forefront of research on the brain and other elements of the nervous system is the National Institute of Neurological Disorders and Stroke (NINDS), which conducts and supports scientific studies in the United States and around the world. This fact sheet is a basic introduction to the human brain. It may help you understand how the healthy brain works, how to keep it healthy, and what happens when the brain is diseased or dysfunctional. <top> The Architecture of the Brain The brain is like a committee of experts. All the parts of the brain work together, but each part has its own special properties. The brain can be divided into three basic units: the forebrain, the midbrain, and the hindbrain. The hindbrain includes the upper part of the spinal cord, the brain stem, and a wrinkled ball of tissue called the cerebellum (1). The hindbrain controls the body’s vital functions such as respiration and heart rate. The cerebellum coordinates movement and is involved in learned rote movements. When you play the piano or hit a tennis ball you are activating the cerebellum. The uppermost part of the brainstem is the midbrain, which controls some reflex actions and is part of the circuit involved in the control of eye movements and other voluntary movements. The forebrain is the largest and most highly developed part of the human brain: it consists primarily of the cerebrum (2) and the structures hidden beneath it (see "The Inner Brain"). When people see pictures of the brain it is usually the cerebrum that they notice. The cerebrum sits at the topmost part of the brain and is the source of intellectual activities. It holds your memories, allows you to plan, enables you to imagine and think. It allows you to recognize friends, read books, and play games. The cerebrum is split into two halves (hemispheres) by a deep fissure. Despite the split, the two cerebral hemispheres communicate with each other through a thick tract of nerve fibers that lies at the base of this fissure. Although the two hemispheres seem to be mirror images of each other, they are different. For instance, the ability to form words seems to lie primarily in the left hemisphere, while the right hemisphere seems to control many abstract reasoning skills. For some as-yet-unknown reason, nearly all of the signals from the brain to the body and vice-versa cross over on their way to and from the brain. This means that the right cerebral hemisphere primarily controls the left side of the body and the left hemisphere primarily controls the right side. When one side of the brain is damaged, the opposite side of the body is affected. For example, a stroke in the right hemisphere of the brain can leave the left arm and leg paralyzed. The Forebrain ------- The Midbrain -------- The Hindbrain
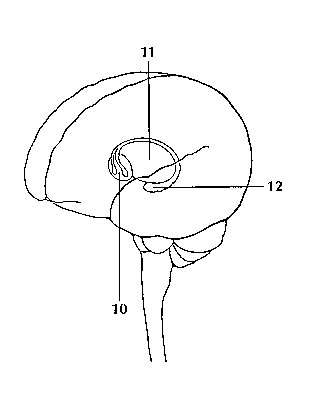
<top> The Geography of Thought Each cerebral hemisphere can be divided into sections, or lobes, each of which specializes in different functions. To understand each lobe and its specialty we will take a tour of the cerebral hemispheres, starting with the two frontal lobes (3), which lie directly behind the forehead. When you plan a schedule, imagine the future, or use reasoned arguments, these two lobes do much of the work. One of the ways the frontal lobes seem to do these things is by acting as short-term storage sites, allowing one idea to be kept in mind while other ideas are considered. In the rearmost portion of each frontal lobe is a motor area (4), which helps control voluntary movement. A nearby place on the left frontal lobe called Broca’s area (5) allows thoughts to be transformed into words. When you enjoy a good meal—the taste, aroma, and texture of the food—two sections behind the frontal lobes called the parietal lobes (6) are at work. The forward parts of these lobes, just behind the motor areas, are the primary sensory areas (7). These areas receive information about temperature, taste, touch, and movement from the rest of the body. Reading and arithmetic are also functions in the repertoire of each parietal lobe. As you look at the words and pictures on this page, two areas at the back of the brain are at work. These lobes, called the occipital lobes (8), process images from the eyes and link that information with images stored in memory. Damage to the occipital lobes can cause blindness. The last lobes on our tour of the cerebral hemispheres are the temporal lobes (9), which lie in front of the visual areas and nest under the parietal and frontal lobes. Whether you appreciate symphonies or rock music, your brain responds through the activity of these lobes. At the top of each temporal lobe is an area responsible for receiving information from the ears. The underside of each temporal lobe plays a crucial role in forming and retrieving memories, including those associated with music. Other parts of this lobe seem to integrate memories and sensations of taste, sound, sight, and touch. <top> The Cerebral Cortex Coating the surface of the cerebrum and the cerebellum is a vital layer of tissue the thickness of a stack of two or three dimes. It is called the cortex, from the Latin word for bark. Most of the actual information processing in the brain takes place in the cerebral cortex. When people talk about "gray matter" in the brain they are talking about this thin rind. The cortex is gray because nerves in this area lack the insulation that makes most other parts of the brain appear to be white. The folds in the brain add to its surface area and therefore increase the amount of gray matter and the quantity of information that can be processed. <top> The Inner Brain Deep within the brain, hidden from view, lie structures that are the gatekeepers between the spinal cord and the cerebral hemispheres. These structures not only determine our emotional state, they also modify our perceptions and responses depending on that state, and allow us to initiate movements that you make without thinking about them. Like the lobes in the cerebral hemispheres, the structures described below come in pairs: each is duplicated in the opposite half of the brain. The hypothalamus (10), about the size of a pearl, directs a multitude of important functions. It wakes you up in the morning, and gets the adrenaline flowing during a test or job interview. The hypothalamus is also an important emotional center, controlling the molecules that make you feel exhilarated, angry, or unhappy. Near the hypothalamus lies the thalamus (11), a major clearinghouse for information going to and from the spinal cord and the cerebrum. An arching tract of nerve cells leads from the hypothalamus and the thalamus to the hippocampus (12). This tiny nub acts as a memory indexer—sending memories out to the appropriate part of the cerebral hemisphere for long-term storage and retrieving them when necessary. The basal ganglia (not shown) are clusters of nerve cells surrounding the thalamus. They are responsible for initiating and integrating movements. Parkinson’s disease, which results in tremors, rigidity, and a stiff, shuffling walk, is a disease of nerve cells that lead into the basal ganglia.
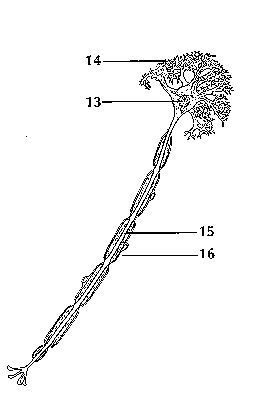
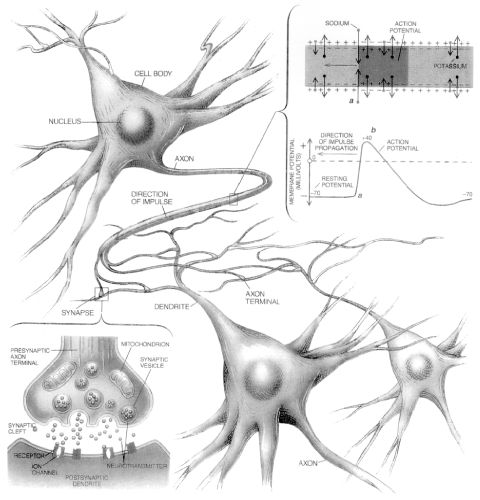
<top> Making Connections The brain and the rest of the nervous system are composed of many different types of cells, but the primary functional unit is a cell called the neuron. All sensations, movements, thoughts, memories, and feelings are the result of signals that pass through neurons. Neurons consist of three parts. The cell body (13) contains the nucleus, where most of the molecules that the neuron needs to survive and function are manufactured. Dendrites (14) extend out from the cell body like the branches of a tree and receive messages from other nerve cells. Signals then pass from the dendrites through the cell body and may travel away from the cell body down an axon (15) to another neuron, a muscle cell, or cells in some other organ. The neuron is usually surrounded by many support cells. Some types of cells wrap around the axon to form an insulating sheath (16). This sheath can include a fatty molecule called myelin, which provides insulation for the axon and helps nerve signals travel faster and farther. Axons may be very short, such as those that carry signals from one cell in the cortex to another cell less than a hair’s width away. Or axons may be very long, such as those that carry messages from the brain all the way down the spinal cord.
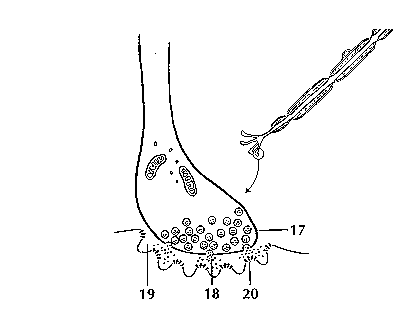
Scientists have learned a great deal about neurons by studying the synapse—the place where a signal passes from the neuron to another cell. When the signal reaches the end of the axon it stimulates tiny sacs (17). These sacs release chemicals known as neurotransmitters (18) into the synapse (19). The neurotransmitters cross the synapse and attach to receptors (20) on the neighboring cell. These receptors can change the properties of the receiving cell. If the receiving cell is also a neuron, the signal can continue the transmission to the next cell.
<top> Some Key Neurotransmitters at Work Acetylcholine is called an excitatory neurotransmitter because it generally makes cells more excitable. It governs muscle contractions and causes glands to secrete hormones. Alzheimer’s disease, which initially affects memory formation, is associated with a shortage of acetylcholine. GABA (gamma-aminobutyric acid) is called an inhibitory neurotransmitter because it tends to make cells less excitable. It helps control muscle activity and is an important part of the visual system. Drugs that increase GABA levels in the brain are used to treat epileptic seizures and tremors in patients with Huntington’s disease. Serotonin is an inhibitory neurotransmitter that constricts blood vessels and brings on sleep. It is also involved in temperature regulation. Dopamine is an inhibitory neurotransmitter involved in mood and the control of complex movements. The loss of dopamine activity in some portions of the brain leads to the muscular rigidity of Parkinson’s disease. Many medications used to treat behavioral disorders work by modifying the action of dopamine in the brain. <top> Neurological Disorders When the brain is healthy it functions quickly and automatically. But when problems occur, the results can be devastating. Some 50 million people in this country—one in five—suffer from damage to the nervous system. The NINDS supports research on more than 600 neurological diseases. Some of the major types of disorders include: neurogenetic diseases (such as Huntington’s disease and muscular dystrophy), developmental disorders (such as cerebral palsy), degenerative diseases of adult life (such as Parkinson’s disease and Alzheimer’s disease), metabolic diseases (such as Gaucher’s disease), cerebrovascular diseases (such as stroke and vascular dementia), trauma (such as spinal cord and head injury), convulsive disorders (such as epilepsy), infectious diseases (such as AIDS dementia), and brain tumors. <top> The National Institute of Neurological Disorders and Stroke Since its creation by Congress in 1950, the NINDS has grown to become the leading supporter of neurological research in the United States. Most research funded by the NINDS is conducted by scientists in public and private institutions such as universities, medical schools, and hospitals. Government scientists also conduct a wide array of neurological research in the more than 20 laboratories and branches of the NINDS itself. This research ranges from studies on the structure and function of single brain cells to tests of new diagnostic tools and treatments for those with neurological disorders. For information on other neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN
NIH Publication No.01-3440a Last updated May 01, 2007 |
The
Life and Death of a NeuronTable of Contents
IntroductionUntil recently, most neuroscientists thought we were born with all the neurons we were ever going to have. As children we might produce some new neurons to help build the pathways - called neural circuits - that act as information highways between different areas of the brain. But scientists believed that once a neural circuit was in place, adding any new neurons would disrupt the flow of information and disable the brain’s communication system. In 1962, scientist Joseph Altman challenged this belief when he saw evidence of neurogenesis (the birth of neurons) in a region of the adult rat brain called the hippocampus. He later reported that newborn neurons migrated from their birthplace in the hippocampus to other parts of the brain. In 1979, another scientist, Michael Kaplan, confirmed Altman’s findings in the rat brain, and in 1983 he found neural precursor cells in the forebrain of an adult monkey. These discoveries about neurogenesis in the adult brain were surprising to other researchers who didn’t think they could be true in humans. But in the early 1980s, a scientist trying to understand how birds learn to sing suggested that neuroscientists look again at neurogenesis in the adult brain and begin to see how it might make sense. In a series of experiments, Fernando Nottebohm and his research team showed that the numbers of neurons in the forebrains of male canaries dramatically increased during the mating season. This was the same time in which the birds had to learn new songs to attract females. Why did these bird brains add neurons at such a critical time in learning? Nottebohm believed it was because fresh neurons helped store new song patterns within the neural circuits of the forebrain, the area of the brain that controls complex behaviors. These new neurons made learning possible. If birds made new neurons to help them remember and learn, Nottebohm thought the brains of mammals might too. Other scientists believed these findings could not apply to mammals, but Elizabeth Gould later found evidence of newborn neurons in a distinct area of the brain in monkeys, and Fred Gage and Peter Eriksson showed that the adult human brain produced new neurons in a similar area. For some neuroscientists, neurogenesis in the adult brain is still an unproven theory. But others think the evidence offers intriguing possibilities about the role of adult-generated neurons in learning and memory.
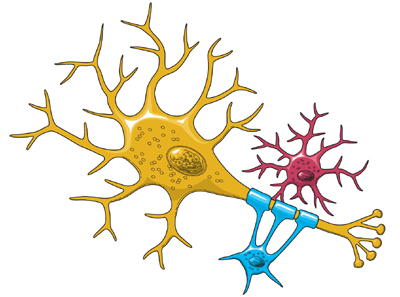
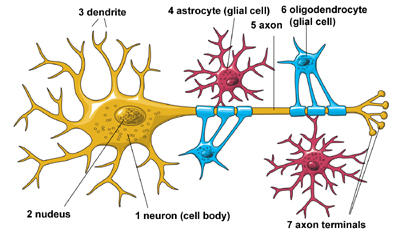
The Architecture of the NeuronThe central nervous system (which includes the brain and spinal cord) is made up of two basic types of cells: neurons (1) and glia (4) & (6). Glia outnumber neurons by a substantial amount -- some scientists have estimated it to be as large as nine to one -- but in spite of their smaller numbers, neurons are the key players in the brain. Neurons are information messengers. They use electrical impulses and chemical signals to transmit information between different areas of the brain, and between the brain and the rest of the nervous system. Everything we think and feel and do would be impossible without the work of neurons and their support cells, the glial cells called astrocytes (4) and oligodendrocytes (6). Neurons have three basic parts: a cell body and two extensions called an axon (5) and a dendrite (3). Within the cell body is a nucleus (2), which controls the cell’s activities and contains the cell’s genetic material. The axon looks like a long tail and transmits messages from the cell. Dendrites look like the branches of a tree and receive messages for the cell. Neurons communicate with each other by sending chemicals, called neurotransmitters, across a tiny space, called a synapse, between the axons and dendrites of adjacent neurons.
There are three classes of neurons:
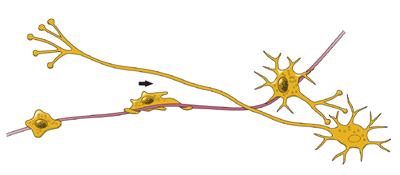
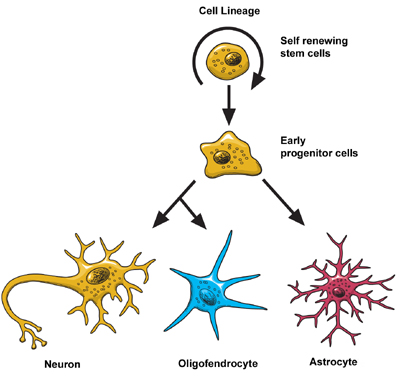
Scientists think that neurons are the most diverse kind of cell in the body. Within these three classes of neurons are hundreds of different types, each with specific message-carrying abilities. How these neurons communicate with each other by making connections is what makes each of us unique in how we think, and feel, and act. BirthThe extent to which new neurons are generated in the brain is a controversial subject among neuroscientists. Although the majority of neurons are already present in our brains by the time we are born, there is evidence to support that neurogenesis (the scientific word for the birth of neurons) is a lifelong process. Neurons are born in areas of the brain that are rich in concentrations of neural precursor cells (also called neural stem cells). These cells have the potential to generate most, if not all, of the different types of neurons and glia found in the brain. Neuroscientists have observed how neural precursor cells behave in the laboratory. Although this may not be exactly how these cells behave when they are in the brain, it gives us information about how they could be behaving when they are in the brain’s environment. The science of stem cells is still very new, and could change with additional discoveries, but researchers have learned enough to be able to describe how neural stem cells generate the other cells of the brain. They call it a stem cell’s lineage and it is similar in principle to a family tree. Neural stem cells increase by dividing in two and producing either two new stem cells, or two early progenitor cells, or one of each. When a stem cell divides to produce another stem cell, it is said to self-renew. This new cell has the potential to make more stem cells. When a stem cell divides to produce an early progenitor cell, it is said to differentiate. Differentiation means that the new cell is more specialized in form and function. An early progenitor cell does not have the potential of a stem cell to make many different types of cells. It can only make cells in its particular lineage. Early progenitor cells can self-renew or go in either of two ways. One type will give rise to astrocytes. The other type will ultimately produce neurons or oligodendrocytes. MigrationOnce a neuron is born it has to travel to the place in the brain where it will do its work. How does a neuron know where to go? What helps it get there? Scientists have seen that neurons use at least two different methods to travel:
Not all neurons are successful in their journey. Scientists think that only a third reach their destination. The rest either never differentiate, or die and disappear at some point during the two to three week phase of migration. Some neurons survive the trip, but end up where they shouldn’t be. Mutations in the genes that control migration create areas of misplaced or oddly formed neurons that can cause disorders such as childhood epilepsy or mental retardation. Some researchers suspect that schizophrenia and the learning disorder dyslexia are partly the result of misguided neurons.
DifferentiationOnce a neuron reaches its destination, it has to settle in to work. This final step of differentiation is the least well-understood part of neurogenesis. Neurons are responsible for the transport and uptake of neurotransmitters - chemicals that relay information between brain cells. Depending on its location, a neuron can perform the job of a sensory neuron, a motor neuron, or an interneuron, sending and receiving specific neurotransmitters. In the developing brain, a neuron depends on molecular signals from other cells, such as astrocytes, to determine its shape and location, the kind of transmitter it produces, and to which other neurons it will connect. These freshly born cells establish neural circuits - or information pathways connecting neuron to neuron - that will be in place throughout adulthood. But in the adult brain, neural circuits are already developed and neurons must find a way to fit in. Researchers suspect that astrocytes play a similar role in the adult brain, actively regulating the function and synapse formation of new neurons. As a new neuron settles in, it starts to look like surrounding cells. It develops an axon and dendrites and begins to communicate with its neighbors.
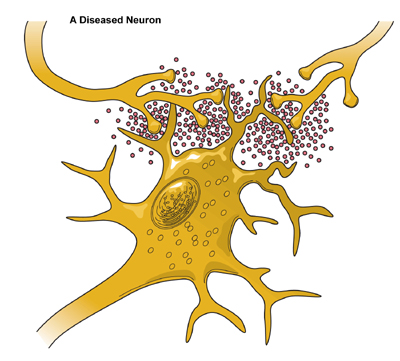
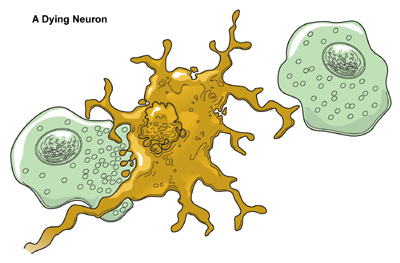
DeathAlthough neurons are the longest living cells in the body, large numbers of them die during migration and differentiation. The lives of some neurons can take abnormal turns. Some diseases of the brain are the result of the unnatural deaths of neurons. - In Parkinson’s disease, neurons that produce the neurotransmitter dopamine die off in the basal ganglia, an area of the brain that controls body movements. The brain can no longer control the body and people shake and jerk in spasms. - In Huntington’s disease, a genetic mutation causes over-production of a neurotransmitter called glutamate, which kills neurons in the basal ganglia. As a result, people twist and writhe uncontrollably. - In Alzheimer’s disease, unusual proteins build up in and around neurons in the neocortex and hippocampus, parts of the brain that control memory. When these neurons die, people lose their capacity to remember and their ability to do everyday tasks. Physical damage to the brain and other parts of the central nervous system can also kill or disable neurons. - Blows to the brain, or the damage caused by a stroke, can kill neurons outright or slowly starve them of the oxygen and nutrients they need to survive. - Spinal cord injury can disrupt communication between the brain and muscles when neurons lose their connection to axons located below the site of injury. These neurons may still live, but they lose their ability to communicate.
Hope Through ResearchScientists hope that by understanding more about the life and death of neurons they can develop new treatments, and possibly even cures, for brain diseases and disorders that affect the lives of millions of Americans. The most current research suggests that neural stem cells can generate many, if not all, of the different types of neurons found in the brain and the nervous system. Learning how to manipulate these stem cells in the laboratory into specific types of neurons could produce a fresh supply of brain cells to replace those that have died or been damaged. Therapies could also be created to take advantage of growth factors and other signaling mechanisms inside the brain that tell precursor cells to make new neurons. This would make it possible to repair, reshape, and renew the brain from within. For information on other neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Prepared
by: NINDS health-related material is provided for information purposes only and does not necessarily represent endorsement by or an official position of the National Institute of Neurological Disorders and Stroke or any other Federal agency. Advice on the treatment or care of an individual patient should be obtained through consultation with a physician who has examined that patient or is familiar with that patient's medical history. All NINDS-prepared information is in the public domain and may be freely copied. Credit to the NINDS or the NIH is appreciated. |
||||||||||||||||||||||||||||||||||||
The
Neuroscience of Mental Health A vast body of research on mental health and, to an even greater extent, on mental illness constitutes the foundation of this Surgeon General’s report. To understand and better appreciate the content of the chapters that follow, readers outside the mental health field may desire some background information. Thus, this chapter furnishes a“primer” on topics that the report addresses. The chapter begins with an overview of research under way today that is focused on the neuroscience of mental health. Modern integrative neuroscience offers a means of linking research on broad“systems level” aspects of brain function with the remarkably detailed tools and findings of molecular biology. The report begins with a discussion of the brain because it is central to what makes us human and provides an understanding of mental health and mental illness. All of human behavior is mediated by the brain. Consider, for example, a memory that most people have from childhood—that of learning to ride a bicycle with the help of a parent or friend. The fear of falling, the anxiety of lack of control, the reassurances of a loved one, and the final liberating experience of mastery and a newly extended universe create an unforgettable combination. For some, the memories are not good ones: falling and being chased by dogs have left marks of anxiety and fear that may last a lifetime. Science is revealing how the skill learning, emotional overtones, and memories of such experiences are put together physically in the brain. The brain and mind are two sides of the same coin. Mind is not possible without the remarkable physical complexity that is built into the brain, but, in addition, the physical complexity of the brain is useless without the sculpting that environment, experience, and thought itself provides. Thus the brain is now known to be physically shaped by contributions from our genes and our experience, working together. This strengthens the view that mental disorders are both caused and can be treated by biological and experiential processes, working together. This understanding has emerged from the breathtaking progress in modern neuroscience that has begun to integrate knowledge from biological and behavioral sciences. An overview of mental illness follows the section on modern integrative brain science. The section highlights topics including symptoms, diagnosis, epidemiology (i.e., research having to do with the distribution and determinants of mental disorders in population groups, including various racial and ethnic minority groups), and cost, all of which are discussed in greater and more pointed detail in the chapters that follow. Etiology is the study of the origins and causes of disease, and that section reviews research that is seeking to define, with ever greater precision, the causes of mental disorders. As will be seen, etiology research examines fundamental biological, behavioral, and sociocultural processes, as well as a necessarily broad array of life events. The section on development of temperament reveals how mental health science has attempted over much of the past century to understand how biological, psychological, and sociocultural factors meld in health as well as in illness. The chapter then reviews research approaches to the prevention and treatment of mental disorders and provides an overview of mental health services and their delivery. Final sections cover the growing influence on the mental health field of the need for attention to cultural diversity, the importance of the consumer movement, and new optimism about recovery from mental illness—that is, the possibility of recovering one’s life. The Neuroscience
of Mental Health1
|
| Excitatory amino acid Glutamate Inhibitory amino acids Monoamines and related neurotransmitters Purine Neuropeptides Opioids Tachykinin Hypothalamic-releasing factors |
Although there are many kinds of receptors with many different signaling functions, we can divide most neurotransmitter receptors into two general classes. One class of neurotransmitter receptor is called a ligand-gated channel, where“ligand” simply means a molecule (i.e., a neurotransmitter) that binds to a receptor. When neurotransmitters interact with this kind of receptor, a pore within the receptor molecule itself is opened and positive or negative charges enter the cell. The entry of positive charge may activate additional ion channels that allow more positive charge to enter. At a certain threshold, this causes a cell to fire an action potential—an electrical event that leads ultimately to the release of neurotransmitter. By definition, therefore, receptors that admit positive charge are excitatory neurotransmitter receptors. The classic excitatory neurotransmitter receptors in the brain utilize the excitatory amino acids glutamate and, to a lesser degree, aspartate as neurotransmitters. Conversely, inhibitory neurotransmitters act by permitting negative charges into the cell, taking the cell farther away from firing. The classic inhibitory neurotransmitters in the brain are the amino acids gamma amino butyric acid, or GABA, and, to a lesser degree, glycine.
Most of the other neurotransmitters in the brain, such as dopamine, serotonin, and norepinephrine, and all of the many neuropeptides constitute the second major class. These are neither precisely excitatory nor inhibitory but rather act to produce complex biochemical changes in the receiving cell. Their receptors do not contain intrinsic ion pores but rather interact with signaling proteins, called“G proteins” found inside the cell membrane. These receptors thus are called G protein-linked receptors. The details are less important than understanding the general scheme. Stimulation of G protein-linked receptors alters the way in which receiving neurons can process subsequent signals from glutamate or GABA. To use a metaphor of a musical instrument, if glutamate, the excitatory neurotransmitter, is puffing wind into a flute or clarinet, it is the modulatory neurotransmitters such as dopamine or serotonin that might be seen as playing the keys and, thus, altering the melody via G protein-linked receptors.
The architecture of these systems drives home this point. The precise brain circuits that carry specific information about the world and that are involved in precise point-to-point communication within the brain use excitatory or inhibitory neurotransmission. Examples of such circuits, which are massively parallel, can be found in the visual and auditory cortex. Overlying this pattern of precise, rapid (timing in the range of milliseconds) neurotransmission are the modulatory systems in the brain that use norepinephrine, serotonin, and dopamine. In each case, the neurotransmitter in question is made by a very small number of nerve cells clustered in a limited number of areas in the brain. Of the hundred billion neurons in the brain, only about 500,000, for example, make dopamine—that is, for every 200,000 cells in the brain, only one makes dopamine. Even fewer make norepinephrine. The cell bodies of the dopamine neurons are clustered in a few brain regions, most importantly, regions deep in the brain, in the midbrain, called the substantia nigra, and the ventral tegmental area. Norepinephrine neurons are made in the nucleus locus coeruleus even farther down in the brain stem in a structure called the pons. Serotonin is made by a somewhat larger number of nuclei but, still, not by many cells. Nuclei called the raphe nuclei spread along the brain stem. While each of these neurotransmitters is made by a small number of neurons with clustered cell bodies, each sends its axons branching throughout the brain, so that in each case a very small number of neurons, which largely appear to fire in unison when excited, influence almost the entire brain. This is not the picture of systems that are communicating precise bits of information about the world but rather are intrinsic modulatory systems that act via other G protein-linked receptors to alter the overall responsiveness of the brain. These neurotransmitters are responsible for brain states such as degree of arousal, ability to pay attention, and for putting emotional color or significance on top of cold cognitive information provided by precise glutaminergic circuits. It is no wonder that these modulatory neurotransmitters and their receptors are critical targets of medications used to treat mental disorders—for example, the antidepressant and antipsychotic drugs—and also are the targets of drugs of abuse.
The preceding paragraphs have illustrated the chemical and anatomic structure of the brain and, in so doing, provided some picture of its complexity as well as some picture of its function. The crowning complexity of the brain, however, is that it is not static. The brain is always changing. People learn so much and have so many distinct types of memory: conscious, episodic memory of the sort that is encoded initially in the hippocampus; memory of motor programs or procedures that are encoded in the striatum; emotional memories that can initiate physiologic and behaviorally adaptive repertoires encoded, for example, in the amygdala; and many other kinds. Every time a person learns something new, whether it is conscious or unconscious, that experience alters the structure of the brain. Thus, neurotransmission in itself not only contains current information but alters subsequent neurotransmission if it occurs with the right intensity and the right pattern. Experience that is salient enough to cause memory creates new synaptic connections, prunes away old ones, and strengthens or weakens existing ones. Similarly, experiences as diverse as stress, substance abuse, or disease can kill neurons, and current data suggest that new neurons continue to develop even in adult brains, where they help to incorporate new memories. The end result is that information is now routed over an altered circuit. Many of these changes are long-lived, even permanent. It is in this way that a person can look back 10 or 20 or 50 years and remember family, a home or school room, or friends. The general theme is that to really understand the kind of memory—indeed, any brain function—one must think at least at two levels: one, the level of molecular and cellular alterations that are responsible for remodeling synapses, and, two, the level of information content and behavior which circuits and synapses serve.
To summarize this section, scientists are truly beginning to learn about the structure and function of the brain. Its awe-inspiring complexity is fully consistent with the fact that it supports all behavior and mental life. Implied in the foregoing, is the fact that brains are built not only by genes—and again, it is the lion’s share of the 80,000 or so human genes that are involved in building a structure so complex as the brain. Genes are not by themselves the whole story. Brains are built and changed through life through the interaction of genes with environment, including experience. It is true that a set of genes might create repetitive multiples of one type of unit, yet the brain appears far more complex than that. It stands to reason that if 50,000 or 60,000 genes are involved in building a brain that may have 100 trillion or a quadrillion synapses, additional information is needed, and that information comes from the environment. It is this fundamental realization that is beginning to permit an understanding of how treatment of mental disorders works—whether in the form of a somatic intervention such as a medication, or a psychological“talk” therapy—by actually changing the brain.
There are many exciting developments in brain science. Of great relevance to the study of mental function and mental illness is the ability to image the activity of the living human brain with technologies developed in recent decades, such as positron emission tomography scanning or functional magnetic resonance imaging. Such approaches can exploit surrogates of neuronal firing such as blood flow and blood oxygenation to provide maps of activity. As science learns more about brain circuitry and learns more from cognitive and affective neuroscience about how to activate and examine the function of particular brain circuits, differences between health and illness in the function of particular circuits certainly will become evident. We will be able to see the action of psychotropic drugs and, perhaps most exciting, we will be able to see the impact of that special kind of learning called psychotherapy, which works after all because it works on the brain.
Different brain chemicals, brain receptors, and brain structures will come up in the discussion of particular illnesses throughout this document. This section is meant to provide a panoramic, not a detailed, introduction and also to provide certain overarching lessons. When something is referred to as biological or brain-based, that is not shorthand for saying it is genetic and, thus, predetermined; similarly, references to “psychological” or even“social” phenomena do not exclude biological processes. The brain is the great integrator, bringing together genes and environment. The study of the brain requires reducing problems initially to bite-sized bits that will allow investigators to learn something, but ultimately, the agenda of neuroscience is not reductionist; the goal is to understand behavior, not to put blinders on and try to explain it away. As the foregoing discussion illustrates, the brain also is complex. Thus, having a disease that affects one or even many critical circuits does not overthrow, except in extreme cases, such as advanced Alzheimer’s disease, all aspects of a person. Typically, people retain their personality and, in most cases, their ability to take responsibility for themselves.
In retrospect, early biological models of the mind seem impoverished and deterministic—for example, models that held that “levels” of a neurotransmitter such as serotonin in the brain were the principal influence on whether one was depressed or aggressive. Neuroscience is far beyond that now, working to integrate information coming “bottom-up” from genes and molecules and cells, with information flowing“top-down” from interactions with the environment and experience to the internal workings of the mind and its neuronal circuits. Ultimately, however, the goal is not only human self-understanding. In knowing eventually precisely what goes wrong in what circuits and what synapses and with what chemical signals, the hope is to develop treatments with greater effectiveness and with fewer side effects. Indeed, as the following chapters indicate, the hope is for cures and ultimately for prevention. There is every reason to hope that as our science progresses, we will achieve those goals.
Development
Stress
and the Developing Brain
Download the pdf file: |
Excessive
Stress Disrupts the Architecture of the Developing Brain.
Download
the pdf file: Used
with express permission from the |
The
Teenage Brain: a work in progress
Part 1: First, download the pdf file: The Teenage Brain: a work in progress
Part 2: Attack
of the Teenage Brain!!
From the pages of medical journals to feature stories on the network news, there's been a swell of media coverage the past few years concerning "the teenage brain." Despite sounding like the title of Hollywood's latest horror-movie blockbuster, the phrase actually refers to recent neurological research on adolescent brain chemistry. To the surprise of practically no one not wearing a lab coat, it's finally been demonstrated scientifically that the teenage brain is different from that of a mature adult. According to the data, these differences explain the average teen's inclination to stay up late, sleep until noon, and exhibit extreme mood swings (for example, from sullen and defiant to really sullen and defiant). Some researchers have even blamed these brain differences for the adolescent's devotion to high-decibel music, insistence on low-decibel mumbling, and willingness to stand in line for hours to see the midnight showing of Watchmen. As soon as these results made national headlines, the usual social pundits weighed in: this new research, they claimed, clearly suggested that we should ban teen driving and even raise the voting age. After all, we now had proof positive that today's teens are simply too erratic to be entrusted with such responsibilities. This may be. But what about the mid-life brain? Perhaps the next time we embark on exhaustive, heavily-funded research into what's inside the human skull, we should focus our efforts on the average middle-aged person. Because if my friends and I are at all representative, I'd argue that whatever's going on in our collective brains is equally suspect. Though not without good reason. We're in the middle of an economic melt-down, deluged with more-bad-news-updates constantly by the media. Not to mention the Middle East crisis, global warming, and daily bulletins about the life and travails of the OctoMom. Most adults I know are over-worked, over-stressed and generally overwhelmed by their ongoing struggles with careers, child-rearing, and relationships. They're forgetful, obsessed with their health (popping pills to an extent no teenager would even contemplate), envious of their neighbors and always---always---sleep-deprived. Frankly, even on a good day, our brains are nothing to write home about. It's everything we can do to keep our complicated, must-have Starbucks coffee orders straight in our heads. I think it's too easy to blame all this on brain chemistry. The truth is, life is hard, no matter how old you are. Whether you're worried about making the track team or paying the mortgage; about fitting in with the cool kids or impressing your new boss, it's all about trying to cope. Granted,
your average teen's coping mechanisms rarely extend beyond
junk food and video games. And, amidst all this, compulsively checking e-mails and sending text messages on their cellphones (while nursing fantasies of winning the Lottery or running off to Tahiti with the office manager). Let's face it, teens have just two basic goals: having sex and getting into a good college. Both pretty laudable and straightforward aims, especially when compared with the confusing and relentless demands of contemporary life with which adults have to contend. It's no wonder, then, that at the end of a hard day, most adults just want to collapse on the sofa and channel-surf. Sartre once said that the state of man is incomprehension and rage. Okay, maybe he was a bit of a Gloomy Gus. But isn't the bewilderment and struggle to which he alludes true at times for all of us, particularly at certain crucial stages in our life? As a psychotherapist, I see daily the unfortunate consequences of assigning a diagnostic label to practically every kind of behavior under the sun. Instead, we need to remember that people are too complex to fit neatly into categories. And that includes teenage people. In fact, before we start debating whether teens should be allowed to drive and vote, we'd better be able to defend letting us adults do so. It's not as if our record in either of these endeavors is anything to brag about. (Okay, we got one right with Obama, but still...) My point is, I think we should give kids a break. They're not responsible for the way their brains develop, any more than they are for the world in which they have to grow up. If anything, the latter is a result of brains much older, and supposedly wiser, than theirs. Formerly
a Hollywood screenwriter (My Favorite Year; Welcome Back, Kotter,
etc.), Dennis Palumbo, MA, MFT is now a licensed psychotherapist
in private practice, specializing in creative issues. His newest
book, a collection of mystery short stories, is called From
Crime to Crime. For more information, please visit his website
at www.dennispalumbo.com |
Specific Neurological Conditions
| Amyotrophic
Lateral Sclerosis
What is amyotrophic
lateral sclerosis?
Who gets ALS? What are the symptoms? How is ALS diagnosed? What causes ALS? How is ALS treated? What research is being done? How Can I Help Research? Where can I get more information? What is amyotrophic lateral sclerosis?Amyotrophic lateral sclerosis (ALS), sometimes called Lou Gehrig's disease, is a rapidly progressive, invariably fatal neurological disease that attacks the nerve cells (neurons) responsible for controlling voluntary muscles. The disease belongs to a group of disorders known as motor neuron diseases, which are characterized by the gradual degeneration and death of motor neurons. Motor neurons are nerve cells located in the brain, brainstem, and spinal cord that serve as controlling units and vital communication links between the nervous system and the voluntary muscles of the body. Messages from motor neurons in the brain (called upper motor neurons) are transmitted to motor neurons in the spinal cord (called lower motor neurons) and from them to particular muscles. In ALS, both the upper motor neurons and the lower motor neurons degenerate or die, ceasing to send messages to muscles. Unable to function, the muscles gradually weaken, waste away (atrophy), and twitch (fasciculations) . Eventually, the ability of the brain to start and control voluntary movement is lost. ALS causes weakness with a wide range of disabilities (see section titled "What are the symptoms?"). Eventually, all muscles under voluntary control are affected, and patients lose their strength and the ability to move their arms, legs, and body. When muscles in the diaphragm and chest wall fail, patients lose the ability to breathe without ventilatory support. Most people with ALS die from respiratory failure, usually within 3 to 5 years from the onset of symptoms. However, about 10 percent of ALS patients survive for 10 or more years. Although the disease usually does not impair a person's mind or intelligence, several recent studies suggest that some ALS patients may have alterations in cognitive functions such as depression and problems with decision-making and memory. ALS does not affect a person's ability to see, smell, taste, hear, or recognize touch. Patients usually maintain control of eye muscles and bladder and bowel functions, although in the late stages of the disease most patients will need help getting to and from the bathroom. Who gets ALS?As many as 20,000 Americans have ALS, and an estimated 5,000 people in the United States are diagnosed with the disease each year. ALS is one of the most common neuromuscular diseases worldwide, and people of all races and ethnic backgrounds are affected. ALS most commonly strikes people between 40 and 60 years of age, but younger and older people also can develop the disease. Men are affected more often than women. In 90 to 95 percent of all ALS cases, the disease occurs apparently at random with no clearly associated risk factors. Patients do not have a family history of the disease, and their family members are not considered to be at increased risk for developing ALS. About 5 to 10 percent of all ALS cases are inherited. The familial form of ALS usually results from a pattern of inheritance that requires only one parent to carry the gene responsible for the disease. About 20 percent of all familial cases result from a specific genetic defect that leads to mutation of the enzyme known as superoxide dismutase 1 (SOD1). Research on this mutation is providing clues about the possible causes of motor neuron death in ALS. Not all familial ALS cases are due to the SOD1 mutation, therefore other unidentified genetic causes clearly exist. What are the symptoms?The onset of ALS may be so subtle that the symptoms are frequently overlooked. The earliest symptoms may include twitching, cramping, or stiffness of muscles; muscle weakness affecting an arm or a leg; slurred and nasal speech; or difficulty chewing or swallowing. These general complaints then develop into more obvious weakness or atrophy that may cause a physician to suspect ALS. The parts of the body affected by early symptoms of ALS depend on which muscles in the body are damaged first. In some cases, symptoms initially affect one of the legs, and patients experience awkwardness when walking or running or they notice that they are tripping or stumbling more often. Some patients first see the effects of the disease on a hand or arm as they experience difficulty with simple tasks requiring manual dexterity such as buttoning a shirt, writing, or turning a key in a lock. Other patients notice speech problems. Regardless of the part of the body first affected by the disease, muscle weakness and atrophy spread to other parts of the body as the disease progresses. Patients have increasing problems with moving, swallowing (dysphagia), and speaking or forming words (dysarthria). Symptoms of upper motor neuron involvement include tight and stiff muscles (spasticity) and exaggerated reflexes (hyperreflexia) including an overactive gag reflex. An abnormal reflex commonly called Babinski's sign (the large toe extends upward as the sole of the foot is stimulated in a certain way) also indicates upper motor neuron damage. Symptoms of lower motor neuron degeneration include muscle weakness and atrophy, muscle cramps, and fleeting twitches of muscles that can be seen under the skin (fasciculations). To be diagnosed with ALS, patients must have signs and symptoms of both upper and lower motor neuron damage that cannot be attributed to other causes. Although the sequence of emerging symptoms and the rate of disease progression vary from person to person, eventually patients will not be able to stand or walk, get in or out of bed on their own, or use their hands and arms. Difficulty swallowing and chewing impair the patient's ability to eat normally and increase the risk of choking. Maintaining weight will then become a problem. Because the disease usually does not affect cognitive abilities, patients are aware of their progressive loss of function and may become anxious and depressed. A small percentage of patients may experience problems with memory or decision-making, and there is growing evidence that some may even develop a form of dementia. Health care professionals need to explain the course of the disease and describe available treatment options so that patients can make informed decisions in advance. In later stages of the disease, patients have difficulty breathing as the muscles of the respiratory system weaken. Patients eventually lose the ability to breathe on their own and must depend on ventilatory support for survival. Patients also face an increased risk of pneumonia during later stages of ALS. How is ALS diagnosed?No one test can provide a definitive diagnosis of ALS, although the presence of upper and lower motor neuron signs in a single limb is strongly suggestive. Instead, the diagnosis of ALS is primarily based on the symptoms and signs the physician observes in the patient and a series of tests to rule out other diseases. Physicians obtain the patient's full medical history and usually conduct a neurologic examination at regular intervals to assess whether symptoms such as muscle weakness, atrophy of muscles, hyperreflexia, and spasticity are getting progressively worse. Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions. One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles. Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV). Specific abnormalities in the NCV results may suggest, for example, that the patient has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS. The physician may order magnetic resonance imaging (MRI), a noninvasive procedure that uses a magnetic field and radio waves to take detailed images of the brain and spinal cord. Although these MRI scans are often normal in patients with ALS, they can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, a herniated disk in the neck, syringomyelia, or cervical spondylosis. Based on the patient's symptoms and findings from the examination and from these tests, the physician may order tests on blood and urine samples to eliminate the possibility of other diseases as well as routine laboratory tests. In some cases, for example, if a physician suspects that the patient may have a myopathy rather than ALS, a muscle biopsy may be performed. Infectious diseases such as human immunodeficiency virus (HIV), human T-cell leukemia virus (HTLV), and Lyme disease can in some cases cause ALS-like symptoms. Neurological disorders such as multiple sclerosis, post-polio syndrome, multifocal motor neuropathy, and spinal muscular atrophy also can mimic certain facets of the disease and should be considered by physicians attempting to make a diagnosis. Because of the prognosis carried by this diagnosis and the variety of diseases or disorders that can resemble ALS in the early stages of the disease, patients may wish to obtain a second neurological opinion. What causes ALS?The cause of ALS is not known, and scientists do not yet know why ALS strikes some people and not others. An important step toward answering that question came in 1993 when scientists supported by the National Institute of Neurological Disorders and Stroke (NINDS) discovered that mutations in the gene that produces the SOD1 enzyme were associated with some cases of familial ALS. This enzyme is a powerful antioxidant that protects the body from damage caused by free radicals. Free radicals are highly reactive molecules produced by cells during normal metabolism. If not neutralized, free radicals can accumulate and cause random damage to the DNA and proteins within cells. Although it is not yet clear how the SOD1 gene mutation leads to motor neuron degeneration, researchers have theorized that an accumulation of free radicals may result from the faulty functioning of this gene. In support of this, animal studies have shown that motor neuron degeneration and deficits in motor function accompany the presence of the SOD1 mutation. Studies also have focused on the role of glutamate in motor neuron degeneration. Glutamate is one of the chemical messengers or neurotransmitters in the brain. Scientists have found that, compared to healthy people, ALS patients have higher levels of glutamate in the serum and spinal fluid. Laboratory studies have demonstrated that neurons begin to die off when they are exposed over long periods to excessive amounts of glutamate. Now, scientists are trying to understand what mechanisms lead to a buildup of unneeded glutamate in the spinal fluid and how this imbalance could contribute to the development of ALS. Autoimmune responses—which occur when the body's immune system attacks normal cells—have been suggested as one possible cause for motor neuron degeneration in ALS. Some scientists theorize that antibodies may directly or indirectly impair the function of motor neurons, interfering with the transmission of signals between the brain and muscles. In searching for the cause of ALS, researchers have also studied environmental factors such as exposure to toxic or infectious agents. Other research has examined the possible role of dietary deficiency or trauma. However, as of yet, there is insufficient evidence to implicate these factors as causes of ALS. Future research may show that many factors, including a genetic predisposition, are involved in the development of ALS. How is ALS treated?No cure has yet been found for ALS. However, the Food and Drug Administration (FDA) has approved the first drug treatment for the disease—riluzole (Rilutek). Riluzole is believed to reduce damage to motor neurons by decreasing the release of glutamate. Clinical trials with ALS patients showed that riluzole prolongs survival by several months, mainly in those with difficulty swallowing. The drug also extends the time before a patient needs ventilation support. Riluzole does not reverse the damage already done to motor neurons, and patients taking the drug must be monitored for liver damage and other possible side effects. However, this first disease-specific therapy offers hope that the progression of ALS may one day be slowed by new medications or combinations of drugs. Other treatments for ALS are designed to relieve symptoms and improve the quality of life for patients. This supportive care is best provided by multidisciplinary teams of health care professionals such as physicians; pharmacists; physical, occupational, and speech therapists; nutritionists; social workers; and home care and hospice nurses. Working with patients and caregivers, these teams can design an individualized plan of medical and physical therapy and provide special equipment aimed at keeping patients as mobile and comfortable as possible. Physicians can prescribe medications to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm. Drugs also are available to help patients with pain, depression, sleep disturbances, and constipation. Pharmacists can give advice on the proper use of medications and monitor a patient's prescriptions to avoid risks of drug interactions. Physical therapy and special equipment can enhance patients' independence and safety throughout the course of ALS. Gentle, low-impact aerobic exercise such as walking, swimming, and stationary bicycling can strengthen unaffected muscles, improve cardiovascular health, and help patients fight fatigue and depression. Range of motion and stretching exercises can help prevent painful spasticity and shortening (contracture) of muscles. Physical therapists can recommend exercises that provide these benefits without overworking muscles. Occupational therapists can suggest devices such as ramps, braces, walkers, and wheelchairs that help patients conserve energy and remain mobile. ALS patients who have difficulty speaking may benefit from working with a speech therapist. These health professionals can teach patients adaptive strategies such as techniques to help them speak louder and more clearly. As ALS progresses, speech therapists can help patients develop ways for responding to yes-or-no questions with their eyes or by other nonverbal means and can recommend aids such as speech synthesizers and computer-based communication systems. These methods and devices help patients communicate when they can no longer speak or produce vocal sounds. Patients and caregivers can learn from speech therapists and nutritionists how to plan and prepare numerous small meals throughout the day that provide enough calories, fiber, and fluid and how to avoid foods that are difficult to swallow. Patients may begin using suction devices to remove excess fluids or saliva and prevent choking. When patients can no longer get enough nourishment from eating, doctors may advise inserting a feeding tube into the stomach. The use of a feeding tube also reduces the risk of choking and pneumonia that can result from inhaling liquids into the lungs. The tube is not painful and does not prevent patients from eating food orally if they wish. When the muscles that assist in breathing weaken, use of nocturnal ventilatory assistance (intermittent positive pressure ventilation [IPPV] or bilevel positive airway pressure [BIPAP]) may be used to aid breathing during sleep. Such devices artificially inflate the patient's lungs from various external sources that are applied directly to the face or body. When muscles are no longer able to maintain oxygen and carbon dioxide levels, these devices may be used full-time. Patients may eventually consider forms of mechanical ventilation (respirators) in which a machine inflates and deflates the lungs. To be effective, this may require a tube that passes from the nose or mouth to the windpipe (trachea) and for long-term use, an operation such as a tracheostomy, in which a plastic breathing tube is inserted directly in the patient's windpipe through an opening in the neck. Patients and their families should consider several factors when deciding whether and when to use one of these options. Ventilation devices differ in their effect on the patient's quality of life and in cost. Although ventilation support can ease problems with breathing and prolong survival, it does not affect the progression of ALS. Patients need to be fully informed about these considerations and the long-term effects of life without movement before they make decisions about ventilation support. Social workers and home care and hospice nurses help patients, families, and caregivers with the medical, emotional, and financial challenges of coping with ALS, particularly during the final stages of the disease. Social workers provide support such as assistance in obtaining financial aid, arranging durable power of attorney, preparing a living will, and finding support groups for patients and caregivers. Respiratory therapists can help caregivers with tasks such as operating and maintaining respirators, and home care nurses are available not only to provide medical care but also to teach caregivers about giving tube feedings and moving patients to avoid painful skin problems and contractures. Home hospice nurses work in consultation with physicians to ensure proper medication, pain control, and other care affecting the quality of life of patients who wish to remain at home. The home hospice team can also counsel patients and caregivers about end-of-life issues. What research is being done?The National Institute of Neurological Disorders and Stroke, part of the National Institutes of Health, is the Federal Government's leading supporter of biomedical research on ALS. The goals of this research are to find the cause or causes of ALS, understand the mechanisms involved in the progression of the disease, and develop effective treatment. Scientists are seeking to understand the mechanisms that trigger selective motor neurons to degenerate in ALS and to find effective approaches to halt the processes leading to cell death. This work includes studies in animals to identify the means by which SOD1 mutations lead to the destruction of neurons. The excessive accumulation of free radicals, which has been implicated in a number of neurodegenerative diseases including ALS, is also being closely studied. In addition, researchers are examining how the loss of neurotrophic factors may be involved in ALS. Neurotrophic factors are chemicals found in the brain and spinal cord that play a vital role in the development, specification, maintenance, and protection of neurons. Studying how these factors may be lost and how such a loss may contribute to motor neuron degeneration may lead to a greater understanding of ALS and the development of neuroprotective strategies. By exploring these and other possible factors, researchers hope to find the cause or causes of motor neuron degeneration in ALS and develop therapies to slow the progression of the disease. Researchers are also conducting investigations to increase their understanding of the role of programmed cell death or apoptosis in ALS. In normal physiological processes, apoptosis acts as a means to rid the body of cells that are no longer needed by prompting the cells to commit "cell suicide." The critical balance between necessary cell death and the maintenance of essential cells is thought to be controlled by trophic factors. In addition to ALS, apoptosis is pervasive in other chronic neurodegenerative conditions such as Parkinson's disease and Alzheimer's disease and is thought to be a major cause of the secondary brain damage seen after stroke and trauma. Discovering what triggers apoptosis may eventually lead to therapeutic interventions for ALS and other neurological diseases. Scientists have not yet identified a reliable biological marker for ALS—a biochemical abnormality shared by all patients with the disease. Once such a biomarker is discovered and tests are developed to detect the marker in patients, allowing early detection and diagnosis of ALS, physicians will have a valuable tool to help them follow the effects of new therapies and monitor disease progression. NINDS-supported researchers are studying families with ALS who lack the SOD1 mutation to locate additional genes that cause the disease. Identification of additional ALS genes will allow genetic testing useful for diagnostic confirmation of ALS and prenatal screening for the disease. This work with familial ALS could lead to a greater understanding of sporadic ALS as well. Because familial ALS is virtually indistinguishable from sporadic ALS clinically, some researchers believe that familial ALS genes may also be involved in the manifestations of the more common sporadic form of ALS. Scientists also hope to identify genetic risk factors that predispose people to sporadic ALS. Potential therapies for ALS are being investigated in animal models. Some of this work involves experimental treatments with normal SOD1 and other antioxidants. In addition, neurotrophic factors are being studied for their potential to protect motor neurons from pathological degeneration. Investigators are optimistic that these and other basic research studies will eventually lead to treatments for ALS. Results of an NINDS-sponsored phase III randomized, placebo-controlled trial of the drug minocycline to treat ALS were reported in 2007. This study showed that people with ALS who received minocycline had a 25 percent greater rate of decline than those who received the placebo, according to the ALS functional rating scale (ALSFRS-R). How Can I Help Research?The NINDS contributes to the support of the Human Brain and Spinal Fluid Resource Center in Los Angeles. This bank supplies investigators around the world with tissue from patients with neurological and other disorders. Tissue from individuals with ALS is needed to enable scientists to study this disorder more intensely. Prospective donors may contact: Human Brain and Spinal Fluid Resource Center Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 00-916 |
|
Introduction
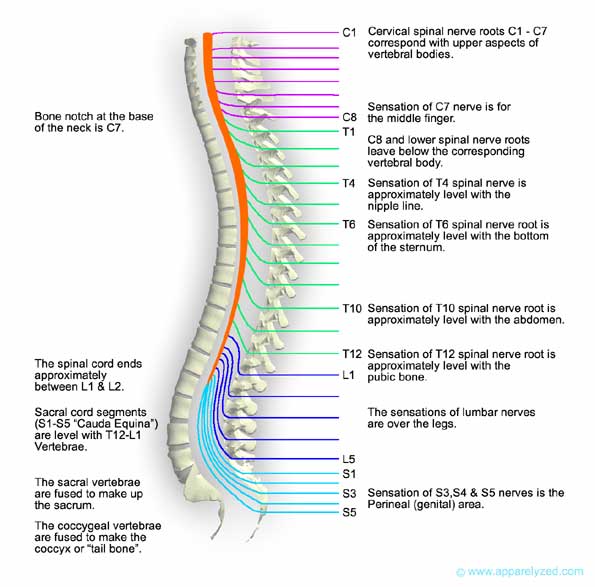
What are Brain and Spinal Cord Tumors? Overview of the brain and spinal cord CNS Tumor FAQS What are benign and malignant tumors? What are primary and metastatic tumors? What causes CNS tumors? Who is at risk? How many people have these tumors? How are tumors graded? What are the possible symptoms? How are CNS tumors diagnosed? How are brain and spinal cord tumors treated? Neurosurgery Radiation Therapy Chemotherapy What is the prognosis? What Research is Being Done? Where can I get more information? Appendix: Some CNS Tumors and Tumor-Related Conditions Glossary IntroductionA diagnosis of a brain or spinal cord tumor brings uncertainty and worry to you and your friends and family. It’s easy to become overwhelmed by a new world of tests, technology, and treatments that you may know little or nothing about. This handbook will give you a better understanding of brain and spinal cord tumors, their treatment options, and the latest research to find safer, more effective ways to diagnose and treat them. You can take the best care of yourself by learning about your diagnosis and discussing it with your doctors. What are Brain and Spinal Cord Tumors?Brain and spinal cord tumors are found in the tissue inside the skull or the bony spinal column, which makes up the central nervous system (CNS). A tumor is a mass of cells that forms a new growth or is present at birth (congenital). Tumors occur when genes that regulate cell growth become damaged or mutated, allowing cells to grow and divide out of control. Tumors can form anywhere in the body. Depending on the type, a growing tumor can kill healthy cells or disrupt their function. It can move or press on sensitive tissue and block the flow of blood and other fluid, causing pain and inflammation. A tumor can also block the normal flow of electricity in the brain or nerve signaling to and from the brain. Some tumors cause no trouble at all. There are more than 120 types of brain and spinal cord tumors. Some are named by the type of cell in which they start (such as glioma) or location (such as meningioma, which form in the lining of the brain and spinal cord). See the Appendix at the end of this guide for a listing of some CNS tumors and tumor-related conditions and the Glossary for specific terms and their meanings. The following overview explains how the CNS works and what happens when a tumor is present. Overview of the brain and spinal cordThe brain has three major parts:
The brain’s two halves, or hemispheres, use nerve cells (neurons) to speak with each other. Each side of the cerebrum controls movement and function on the other side of the body. In addition, each hemisphere has four sections, called lobes, which handle different neurological functions. The frontal lobes manage voluntary movement, such as writing, and let us set and prioritize goals. A frontal lobe tumor can cause changes in personality, intellect, reasoning, and behavior; affect coordination and walking, and cause speech loss. The temporal lobes are linked to perception, memory, and understanding sounds and words. A tumor here might cause speech and hearing problems, blackouts, seizures, or sensations such as a feeling of fear. The parietal lobes let us simultaneously receive and understand sensations such as pressure and pain. A parietal lobe tumor might cause difficulty understanding or speaking words, problems with coordination, seizures, and numbness or weakness on one side of the body. The occipital lobes receive and process light and visual images, and detect motion. An occipital lobe tumor can affect the field of vision, usually on one side of the face, and how we understand written words. Three layers of protective tissue (called the meninges) cover the brain—the thick dura mater (outer layer), the arachnoid (middle), and the pia mater (innermost to the brain). Brain tumors in infants and adults tend to be located in the cerebrum. Brain tumors in children ages 1-12 years are more commonly found in the cerebellum. The spinal cord—an extension of the brain—lies protected inside the bony spinal column. It contains bundles of nerves that carry messages between the brain and other parts of the body, such as instructions from the brain to move an arm or information from the skin that signals pain. A tumor that forms on or near the spinal cord can disrupt communication between the brain and the nerves or restrict the cord's supply of blood. Because the spinal column is narrow, a tumor here—unlike a brain tumor—can cause symptoms on both sides of the body at the same time. Most spinal cord tumors form below the neck. Symptoms generally strike body areas at the same level or at a level below that of the tumor. For example, a tumor midway along the spinal cord (in the thoracic spine) can cause pain that spreads over the chest and gets worse when the individual coughs, sneezes, or lies down. A tumor that grows in the cervical spine can cause pain that seems to come from the neck or arms, and a tumor that grows in the lower, lumbar spine can trigger back or leg pain. The three major groups of spinal cord tumor describe where they are found. Extradural tumors grow between the inner surface of the spinal canal and the tough dura mater. Tumors inside the dura (intradural tumors) are further divided into those outside the spinal cord (extramedullary tumors) and those inside the spinal cord (intramedullary tumors). Other descriptors for spinal cord tumors are intrinsic, meaning the tumor forms inside the spinal cord; and extrinsic, where the tumor forms outside of and presses on the cord as it grows. CNS Tumor FAQSWhat are benign and malignant tumors?No matter where they are located in the body, tumors are classified as benign or malignant. Benign tumors are slow growing, non-cancerous cell masses that have a defined edge and do not spread to other parts of the body. Cells in the tumor are similar to normal cells. Often these tumors can be removed surgically and usually do not recur. Malignant, or cancerous, tumors have cells that look different from normal cells. They can quickly invade surrounding tissue and often have edges that are hard to define, which makes it difficult to remove the entire tumor surgically. What are primary and metastatic tumors?Primary tumors of the CNS are growths that begin in the brain or spinal cord. They can be either malignant or benign and are identified by the types of cells they contain, their location, or both. Most primary CNS tumors occur in adults. Metastatic, or secondary, tumors in the CNS are caused by cancer cells that break away from the primary tumor that developed in a non-CNS part of the body. These tumors are named after the type of cancer that causes them. Metastastic tumors (also called metastases) to the brain occur in about one-fourth of all cancers that develop in other parts of the body, such as cancer of the lung, breast, or kidneys; or melanoma, a form of skin cancer. They are more common than primary tumors and occur more often in adults than in children. Metastatic spine tumors usually form within the bony covering of the spinal column but may also invade the spinal canal from the chest or abdomen. While cancers elsewhere in the body can easily cause tumors inside the brain and spinal cord, CNS tumors rarely spread outside the nervous system. What causes CNS tumors?Researchers really don't know why primary brain and spinal cord tumors develop. Possible causes under investigation include viruses, defective genes, exposure to certain chemicals and other hazardous materials, and immune system disorders. Although smoking, alcohol consumption, and certain dietary habits are associated with some types of cancers, they have not been linked to primary CNS tumors. In a small number of individuals, CNS tumors may result from specific genetic diseases, such as neurofibromatosis and tuberous sclerosis, or exposure to radiation. Non-ionizing radiation (radio waves) from mobile phone use does not increase the risk of developing a brain tumor.1 Brain and spinal cord tumors are not contagious or, at this time, preventable. Who is at risk?Anyone can develop a primary CNS tumor, although the risk is very small. Having one or more of the known risk factors does not guarantee that someone will develop a tumor. Brain tumors occur more often in males than in females and are most common in middle-aged to older persons. They also tend to occur more often in children under age 9 than in other children, and some tumors tend to run in families. Most brain tumors in children are primary tumors. Other risk factors for developing a primary CNS tumor include race (Caucasians are more likely to develop a CNS tumor than other races) and occupation. Workers in jobs that require repeated contact with ionizing radiation or certain chemicals, including those used to manufacture building supplies or plastics and textiles, have a greater chance of developing a brain tumor. How many people have these tumors?More
than 359,000 persons in the
Spinal cord tumors are less common than brain tumors. Although they affect people of all ages, spinal cord tumors are most common in young and middle-aged adults. Nearly 3,200 central nervous system tumors are diagnosed each year in children under age 20.4 How are tumors graded?The generally accepted scale for grading CNS tumors was approved by the World Health Organization in 1993. Grading is based on the tumor’s cellular makeup and location. Tumors may also be classified as low-grade (slowly growing) or high-grade (rapidly growing). Some tumors change grades as they progress, usually to a higher grade, and can become a different type of tumor. The tumor is graded by a pathologist following a biopsy or during surgery. Grade I tumors grow slowly and generally do not spread to other parts of the brain. It is often possible to surgically remove an entire grade I benign tumor, but this type of tumor may be monitored periodically, without further treatment. Grade II tumors also grow slowly, sometimes into surrounding tissue, and can become a higher-grade tumor. Treatment varies according to tumor location and may require chemotherapy, radiation, or surgery followed by close observation. Grade III tumors are malignant and can spread quickly into other CNS tissue. Tumor cells will look different than those in surrounding tissue. Aggressive treatment, often using a combination of chemotherapy, radiation, and/or surgery, is required. Grade IV tumors invade nearby tissue very quickly and are difficult to treat. The cancerous tissue will look very different from surrounding tissue. Aggressive treatment is required. What are the possible symptoms?Brain and spinal cord tumors cause many diverse symptoms, which can make detection tricky. Symptoms depend on tumor type, location, size, and rate of growth. Certain symptoms are quite specific because they result from damage to particular areas of the brain. Symptoms generally develop slowly and worsen as the tumor grows. The most obvious sign of a brain tumor in infants is a rapidly widening head or bulging crown. Common symptoms of a brain tumor in adults include headaches, seizures, problems with balance or coordination, loss of muscle control, hydrocephalus, and changes in personality or behavior. Headaches are the most common symptom of a brain tumor. Headaches may progressively worsen, become more frequent or constant, and recur, often at irregular intervals. Headache pain may worsen when coughing, changing posture, or straining and may be severe upon waking. Seizures can occur, with symptoms that may include convulsions, loss of consciousness, or loss of bladder control. Seizures that first start in adulthood (in someone who has not been in an accident or who had an illness that causes seizures) are a key warning sign of brain tumors. Nausea and vomiting may be more severe in the morning and may accompany headaches. Vision or hearing problems can include blurred or double vision, partial or total loss of vision or hearing, ringing or buzzing sounds, and abnormal eye movements. Personality, behavior, and cognitive changes can include psychotic episodes and problems with speech, language, thinking, and memory. Motor problems can include weakness or paralysis, lack of coordination, or gradual loss of sensation or movement in an arm or leg. A sudden, marked change in handwriting may be a sign of a tumor. Balance problems can include dizziness, trouble with walking, clumsiness, or loss of the normal control of equilibrium. Hydrocephalus and increased intracranial pressure are caused when a tumor blocks the flow of the cerebrospinal fluid (CSF) that surrounds the brain and spinal cord. Symptoms may include headaches, nausea, and vomiting. Other symptoms may include endocrine disorders or abnormal hormonal regulation, difficulty swallowing, facial paralysis and sagging eyelid, fatigue, weakened sense of smell, or disrupted sleep and sleep pattern changes. Common symptoms of a spinal cord tumor include pain, numbness or sensory changes, and motor problems and loss of muscle control. Pain can feel as if it is coming from various parts of the body. Back pain may extend to the hips, legs, feet, and arms. This pain is often constant and may be severe. It is often progressive and can have a burning or aching quality. Numbness or sensory changes can include decreased skin sensitivity to temperature and progressive numbness or a loss of sensation, particularly in the legs. Motor problems and loss of muscle control can include muscle weakness, spasticity (in which the muscles stay stiffly contracted), and impaired bladder and/or bowel control. If untreated, symptoms may worsen to include muscle wasting, decreased muscle strength, an abnormal walking rhythm known as ataxia, and paralysis. Symptoms may spread over various parts of the body when one or more tumors extend over several sections of the spinal cord. How are CNS tumors diagnosed?A doctor, usually a neurologist, oncologist, or neuro-oncologist, can confirm a diagnosis of a brain or spinal cord tumor based on a patient’s symptoms, personal and family medical history, and results of a physical exam and specialized tests and techniques. A neurological exam—the first test—assesses movement and sensory skills, hearing and speech, reflexes, vision, coordination and balance, mental status, and changes in mood or behavior, among other abilities. Some tests require a specialist to perform and analyze results. Diagnostic Imaging The doctor will order one or more imaging techniques that show extremely detailed views of body structures, including tissues, organs, bones, and nerves. If there is a tumor, diagnostic imaging will confirm the diagnosis and help doctors determine the tumor's type, detect swelling and other associated conditions, and, over time, check the results of treatment. See the NINDS publication, “Neurological Diagnostic Tests and Procedures,” for a more complete description of the following tests: Computed Tomography (CT) uses x-rays and a computer to produce fast, detailed cross-sectional images or “slices” of organs, bones, and tissues, including a tumor. It is also good for detecting the buildup of calcium, which causes tissue to harden and can develop into a tumor. Magnetic Resonance Imaging (MRI) uses a computer, radio waves, and a strong magnetic field to produce two-dimensional slices or a detailed three-dimensional model of tissue being scanned. MRI takes longer to perform than does a CT but is more sensitive and gives better pictures of tumors located near bone. Both CT and MRI scans for tumor are usually performed before and after administration of a "contrast" agent (such as a dye) that is given into a vein. Many tumors become much brighter on the scan taken after the contrast is given. Functional MRI (fMRI) creates images of areas of the brain with specific functions such as movement and language. It can assess brain damage from head injury or degenerative disorders, identify and monitor other neurological disorders such as stroke, and show the distance between specific brain functions and tumors in particular areas of the brain. Magnetic Resonance Spectroscopy (MRS) gives doctors a chemical snapshot of tissues being studied. It uses the MRI scanner's magnetic field and radio waves to measure and analyze the chemical make-up of the tissue sample. Positron Emission Tomography (PET) provides computer-generated two- and three-dimensional scans of the brain’s chemical activity and cellular function. PET traces and measures the brain’s use of glucose (sugar, used by the brain for energy) that is attached to small amounts of radioactivity and injected into the bloodstream. Since malignant tissue uses more glucose than normal tissue, it usually shows up on the scan as brighter than surrounding tissue. Single Photon Emission Computed Tomography (SPECT) studies blood flow to tissue. Certain tumors grow new blood vessels to increase their supply of blood and nutrients. A radioactive isotope is injected intravenously and traced as it travels into the skull. A sophisticated computer processes and stacks the data into a detailed three-dimensional image of activity within the brain. Angiography (or arteriogram) can distinguish certain types of tumors that have a characteristic pattern of blood vessels and blood flow. A dye that deflects x-rays is injected into a major blood vessel and a series of x-rays is taken as the dye flows to the brain. In many situations angiography has been replaced by non-invasive tests such as CT and MRI. Testing blood, urine, and other substances can provide clues about the tumor and monitor levels of therapeutic drugs. Additional tests may include an electroencephalogram, or EEG, which monitors brain activity through the skull (tumors can disrupt the normal flow of brain wave activity and cause seizures); CSF analysis, in which a small amount of the cerebrospinal fluid is removed by a special needle and examined for abnormal cells or unusual levels of various compounds that suggest a brain or spinal cord tumor; and magnetoencephalography (MEG), which studies brain function by measuring the magnetic field generated by nerve cells in the brain. CSF fluid analysis should be performed with extreme caution on individuals with very large brain tumors. Diagnosing the distinct type of brain tumor is often difficult. Individuals should consider asking their primary care physician or oncologist for a second opinion, particularly from a neuro-oncologist or neurosurgeon, as there may be new information available and some tumors can change grade or recur. Even a second opinion that confirms the original diagnosis can help people better prepare for their care and treatment. How are brain and spinal cord tumors treated?A specialized team of doctors advises and assists individuals throughout treatment and rehabilitation. These doctors may include: A neurologist is a specialist in nervous system disorders. A medical oncologist is a specialist in cancer. A neuro-oncologist is a neurologist who specializes in nervous system tumors. A neuroradiologist is a doctor trained in reading diagnostic imaging results who specializes in the CNS. A pathologist is a clinical physician who diagnoses diseases of tissues or cells using a variety of laboratory tests. A neurosurgeon is a brain or spinal cord surgeon. A radiation oncologist is a doctor who specializes in using radiation to treat individuals with cancer. This team will recommend a treatment plan based on the tumor's location, type, size and aggressiveness, as well as on the individual’s medical history, age, and general health. Initial treatment for a CNS tumor may involve a variety of drugs, including anticonvulsants to treat seizures, pain medications, steroids or other anti-inflammatory drugs to reduce swelling and improve blood flow, antidepressants to treat anxiety or ease depression that might occur following a tumor diagnosis, and drugs to fight nausea caused by various treatments. Malignant tumors require some form of treatment, while some small benign tumors may need only periodic monitoring. The three standard treatment options for malignant CNS tumors are neurosurgery, radiation therapy, and chemotherapy. Some patients may receive a combination of treatments. NeurosurgerySurgery is usually the first step in treating an accessible tumor—one that can be removed without unacceptable risk of neurological damage. Surgery is aimed at removing all or as much tumor as possible (called resecting or excising) and can often slow worsening of neurological function. An inaccessible or inoperable tumor is one that cannot be removed surgically because of the risk of severe nervous system damage during the operation. These tumors are frequently located deep within the brain or near vital structures such as the brain stem. A biopsy is usually performed to help doctors determine how to treat a tumor. A brain biopsy involves surgically removing a small part of the skull to sample the tumor tissue. Biopsies can sometimes be performed by a needle inserted through a small hole. A small piece of tissue remains in the hollow needle when it is removed from the body. A pathologist will stain and examine the tissue for certain changes that signal cancer and grade it to reflect the degree of malignancy. If the sample is cancerous, the surgeons will remove as much of the tumor as possible. For some primary brain tumors it is not possible to surgically remove all malignant cells. Malignant brain tumors commonly recur from cells that have spread from the original tumor mass into the surrounding brain tissue. In contrast, many benign tumors and secondary metastatic tumors can be completely removed surgically. In some cases, a surgeon may need to insert a shunt into the skull to drain any dangerous buildup of CSF caused by the tumor. A shunt is a flexible plastic tube that is used to divert the flow of CSF from the central nervous system to another part of the body, where it can be absorbed as part of the normal circulatory process. Fortunately, research has led to advances in neurosurgery that make it possible for doctors to completely remove many tumors that were previously thought to be inoperable. These new techniques and tools let neurosurgeons operate within the tight, vulnerable confines of the CNS. Some tools used in the operating room include a surgical microscope, the endoscope (a small viewing tube attached to a video camera), and miniature precision instruments that allow surgery to be performed through a small incision in the brain or spine. Intraoperative MRI uses a special type of MRI to provide a real-time evaluation of the surgery. Constantly updated images that are provided during surgery let doctors see how much tumor material has been removed. Intraoperative MRI can also help doctors choose the best surgical approaches and monitor any complications during surgery. Navigation equipment used in computer-guided, or stereotactic, neurosurgery gives doctors a precise, three-dimensional map of an individual’s spine or brain as the operation progresses. A computer uses pre-operative diagnostic images of the individual to reduce the risk of damage to surrounding tissue. Intraoperative nerve monitoring tests such as evoked potentials use real-time recordings of nerve cell activity to determine the role of specific nerves and monitor brain activity as the surgery progresses. Small electrodes are used to stimulate a nerve and measure its electrical response (or evoked potential). Some surgeries may be done while the individual is awake under monitored anesthesia care, rather than under general anesthesia. This allows doctors to monitor the individual’s speech and motor functions as a tumor is being removed. A possible side effect of surgery is swelling around the site of the tumor, and can be treated with steroids. Bleeding into the tumor site or infection are other serious risks of brain surgery. In the cae of metastatic tumors, doctors usually treat the original cancer. However, if there are only one or two metastases to the brain or if a metastatic tumor causes serious disability or pain, doctors may recommend surgery—even if the original cancer has not been controlled. Surgery may be the beginning and end of your treatment if the biopsy shows a benign tumor. If the tumor is malignant, doctors often recommend additional treatment, including radiation and chemotherapy, or one of several experimental treatments. Radiation TherapyRadiation therapy usually involves repeated doses of x-rays or other forms of radiation to kill cancer cells or keep them from multiplying. When successful, this therapy shrinks the tumor mass but does not actually remove it. Radiation therapy can be used to treat surgically inaccessible tumors or tumor cells that may remain following surgery. Depending on tumor type and stage, radiation treatment might be delivered externally, using focused beams of energy or charged particles that are directed at the tumor, or internally, using a surgically implanted device. The stronger the radiation, the deeper it can penetrate to the target site. Healthy cells may also be damaged by radiation therapy but most are able to repair themselves, while the damaged tumor cells cannot. A radiation oncologist will explain the therapy and how much radiation is needed. Treatment often begins a week after surgery and may continue for several weeks. Depending on tumor type and location, individuals may be able to receive a modified form of therapy to lessen damage to healthy cells and improve the overall treatment. Externally-delivered radiation therapy poses no risk of radioactivity to the patient or family and friends. Types of external radiation therapy include: Whole brain radiation is generally used to shrink multiple cancerous tumors, rather than to target individual tumors. It may be given as the sole form of treatment or in advance of other forms of radiation therapy and microsurgery. Whole brain radiation is generally used for metastatic tumors and rarely for primary tumors. Conventional external beam radiation aims a uniform dose of high-energy radiation at the tumor and surrounding tissue from outside the body. It is used to treat large tumors or those that may have spread into surrounding tissue. Three-dimensional conformal radiotherapy (3D-CRT) uses diagnostic imaging to prepare an accurate, computer-generated three-dimensional image of the tumor and surrounding tissue. The computer then coordinates and sends multiple beams of radiation to the tumor’s exact shape, sparing nearby organs and surrounding tissue. Intensity modulated radiation therapy (IMRT) is similar to 3D-CRT but varies the intensity of the hundreds of individual radiation beams to deliver more precise doses to the tumor or to specific areas within it. IRMT can provide a highly effective dose of radiation to the tumor, with less exposure to surrounding tissue. Hyperfractionation involves giving two or more smaller amounts of radiation a day instead of a larger, single dose. It can deliver more radiation to certain tumors and reduce damage to normal cells. Radiosurgery is usually a one-time treatment involving a large amount of sharply focused radiation that is aimed at the brain tumor. Stereotactic radiosurgery uses computer imaging to direct precisely focused radiation into the tumor from multiple angles. It does not actually cut into the person but, like other forms of radiation therapy, harms a tumor cell’s ability to grow and divide. Stereotactic radiosurgery is commonly used to treat surgically inaccessible tumors. It also may be used at the end of conventional radiation treatment. Two common stereotactic radiosurgery procedures are:
Both procedures may be given on an outpatient basis but an overnight stay in the hospital is often recommended. Proton beam therapy directs a beam of high-energy protons directly at the tumor site, leaving surrounding healthy tissue and organs intact. It is best used to treat tumors that are solid and have not spread to other parts of the body. Proton beam therapy can be used as a stand-alone treatment or in combination with chemotherapy or as follow-up to surgery. Internal tumor radiation therapy, also called brachytherapy or interstitial radiation therapy, involves placing a small amount of radioactive material into or near the tumor (or its cavity, if the tumor has been surgically removed). In most cases, the radiation is inserted at the time of surgery or using imaging and a catheter. The radiation may be left in place for several days and, if more than one treatment is needed, the doctor may leave the catheter in place for a longer period. Individuals may need to be hospitalized for a few days following this procedure as the radiation may extend outside their body and could possibly harm others. The radiation becomes less active each day until it is safe for individuals who have been treated to be around others. Side effects of radiation therapy vary from person to person and are usually temporary. They typically begin about two weeks after treatment starts and may include fatigue, nausea, vomiting, reddened or sore skin in the area receiving treatment, headache, hearing loss, problems with sleep, and hair loss (although the hair usually grows back once the treatment has stopped). Radiation therapy in young children, particularly those children age three or younger, can cause problems with learning, processing information, thinking, and growing. ChemotherapyChemotherapy uses powerful drugs to kill cancer cells or stop them from growing or dividing. These drugs are usually given by pill or injection and travel through the body to the brain, or can be inserted surgically using dissolvable wafers that have been soaked in a chemotherapeutic drug. These wafers slowly release a high concentrate of the drug to kill any remaining malignant cells. Chemotherapy may also be given to kill cancer cells in the spinal column. Chemotherapy is given in cycles to more effectively harm cancer cells and give normal cells time to recover from any damage. The oncologist will base the treatment on the type of cancer, drug(s) to be used, the frequency of administration, and the number of cycles needed. Individuals might receive chemotherapy to shrink the tumor before surgery (called neo-adjuvant therapy), in combination with radiation therapy, or after radiation treatment (called adjuvant therapy) to destroy any remaining cancer cells. Metronomic therapy involves giving continuous low-dose chemotherapy to block mechanisms that stimulate the growth of new blood vessels needed to feed the tumor. Chemotherapy is also used to treat CNS lymphoma and inaccessible tumors or tumors that do not respond to radiation therapy. Not all tumors are vulnerable to the same anticancer drugs, so a person’s treatment may include a combination of drugs. Common CNS chemotherapeutics include temozolomide, carmustine (also called BCNU), lomustine, tamoxifen, carboplatin, methotrexate, procarbazine, and vincristine. Individuals should be sure to discuss all options with their medical team. Side effects of chemotherapy may include hair loss, nausea, digestive problems, reduced bone marrow production, and fatigue. The treatment can also harm normal cells that are growing or dividing at the same time, but these cells usually recover and problems stop once the treatment has ended. Alternative and complementary approaches may help tumor patients better cope with their diagnosis and treatment. Some of these therapies, however, may be harmful if used during or after cancer treatment and should be discussed with a doctor beforehand. Common approaches include nutritional and herbal supplements, vitamins, special diet, and mental or physical techniques to reduce stress. What is the prognosis?Each person is different. Prognosis depends greatly on prompt diagnosis and treatment, the individual’s age and general health, whether the tumor is malignant or benign, tumor size and location, tumor grade, and response to therapy. An individual whose entire tumor has been removed successfully may recover completely. Generally, prognosis is poorer in very young children and in older individuals. Rehabilitation and counseling can help patients and family members better cope with the disorder and improve quality of life. Continued monitoring and long-term follow-up is advised as many tumors resist treatment and tend to recur. Normal tissue and nerves that may have been damaged or traumatized by the tumor or its treatment will need time to heal. Some post-treatment symptoms will disappear over time. Physical therapy can help people regain motor skills, muscle strength, and balance. Some individuals may need to relearn how to swallow or speak if the brain’s cognitive areas have been affected. Occupational therapy can teach people new ways to perform tasks. Supportive care can help people manage any pain and other symptoms. What Research is Being Done?Scientists continue to investigate ways to better understand, diagnose, and treat CNS tumors. Several of today’s treatment regimens were experimental therapies only a decade ago. Current clinical studies of genetic risk factors, environmental causes, and molecular mechanisms of cancers may translate into tomorrow’s treatment of, or perhaps cure for, these tumors. Much
of this work is supported by the National Institutes of Health
(NIH), through the collaborative efforts of its National Institute
of Neurological Disorders and Stroke (NINDS) and National Cancer
Institute (NCI), as well as other agencies within the Federal
government, nonprofit groups, pharmaceutical companies, and private
institutions.
Some of this research is conducted through the collaborative
neuroscience and cancer research community at the NIH or through
research grants to academic centers throughout the
The jointly sponsored NCI-NINDS Neuro-Oncology Branch coordinates research to develop and test the effectiveness and safety of novel therapies for people with CNS tumors. These experimental treatment options may include new drugs, combination therapy, gene therapy, biologic immuno-agents, surgery, and radiation. Information about these trials, and trials involving other disorders, can be accessed at the Federal government’s database of clinical trials, http://clinicaltrials.gov. Part of this research involves creating a comprehensive public database of clinical, molecular, and genetic information on brain tumors to help researchers and clinicians identify and evaluate molecular targets in brain cancer. The joint NCI-NINDS Repository for Molecular Brain Neoplasia Data (REMBRANDT) will hold genetic and molecular analysis of samples from brain tumor patients in NCI-sponsored clinical trials in what is slated to become the largest clinical/genetic corollary study ever conducted on brain tumors. It will also store a wide array of molecular and genetic data regarding all types of brain tumors. Understanding the biology behind these tumors will provide clues to new therapeutic approaches. Researchers are also developing mouse models that mimic human CNS cancers, which may speed discovery of potential treatments in humans. Beyond their efforts to develop needed resources, scientists at the NIH and at universities across the US are are exploring a variety of approaches to treat CNS tumors. These experimental approaches include boosting the immune system to better fight tumor cells, developing therapies that target the tumor cell while sparing normal cells, making improvements in radiation therapy, disabling the tumor using genes attached to viruses, and defining biomarkers that may predict the response of a CNS tumor to a particular treatment. Biological therapy involves enhancing the body’s overall immune response to recognize and fight cancer cells. The immune system is designed to attack foreign substances in the body; since cancer cells aren’t foreign, they usually do not generate much of an immune response. Researchers are using different methods to provoke a strong immune response to tumor cells. Such proteins as interleukin and interferon, both of which are immune system “messengers” called cytokines, and other substances can stimulate and restore the body’s natural response against proteins on the surfaces of cancer cells and thus slow tumor growth. Other therapy uses viruses, T-cells (a major component of immune system function), and other substances to increase immune response and target the tumor cells. Scientists are also experimenting with genetically altered T-cells from the patient’s own body, which are cultured with an antigen and injected directly into the brain following surgery. Biological therapies to fight CNS tumors include vaccines, gene therapy, antibody therapy, and tumor growth factors. Antibodies are proteins that are normally produced by the body to ward off bacteria and viruses. Monoclonal antibodies are multiple copies of a single antibody that act as a homing device to fit one—and only one—type of antigen (a protein on the surface of the tumor cell that stimulates the immune system response). Scientists at the NINDS and elsewhere are linking these antibodies to immunotoxins that seek out tumor cells with a matching antigen, bind to these tumor cells, and deliver their toxin, with minimal damage to surrounding normal cells. Additional approaches include attaching a radioactive substance to the antibodies, which act as targeting agents to deliver radiation to the tumor cell. Gene therapy (also called gene transfer) aims to deliver a suicide gene to the tumor cell. It can also be used to boost the immune system. A gene whose activity can be influenced to kill the cell is integrated into a virus that can cross the blood-brain barrier (an elaborate network of fine blood vessels and cells that filters the blood reaching the CNS) and travel to the tumor. Delivery methods under investigation include viruses and stem cells. Gene therapy may become an important add-on therapy for individuals who do not respond well to other treatments. Scientists are testing the effectiveness of vaccine therapy for a variety of CNS tumors. Vaccine therapy strengthens the immune response by inserting an antigen that the body will attack. Some vaccines target a specific antigen, while others, using the whole tumor cell to make the vaccine, hope to target multiple antigens which the tumor may express. Research has shown the vaccine can be genetically engineered with tumor antigens and injected into the body to induce an immune response, which increases the attack on antigens expressed on the surface of the tumor cell. Experimental CNS tumor vaccines include those designed to stimulate an immune response to a particular protein or antigen, and dendritic cell vaccines (in which blood cells are taken from the body, processed in a laboratory and given back to the patient to break up the tumor cell outer protein into smaller pieces, which increases targeting by immune cells). Findings from NIH-sponsored research on adult CNS tumor development and treatment suggest the immune system and, potentially, infection with the cytomegalovirus may play a critical role in certain tumor-type risk and prognosis. Repeated stimulation of the immune system to counter this herpes virus may serve as a tumor promotion factor. Scientists hope to identify immune factors and develop targeted immunotherapies for this disease. Targeted therapy uses different molecules to reduce tumor gene expression and suppress uncontrolled growth by killing or reducing the production of tumor cells. Of particular interest to scientists is the development of tailored therapeutics—involving a combination of targeted agents—to treat tumors based on tehir genetic make-up. Specifically, molecularly targeted drugs seek out the molecular and cellular changes that convert normal cells into cancer. Many targeted cancer therapies are being tested in animals for use alone or in combination with other cancer treatments, such as chemotherapy. Researchers hope to discover tumor-specific cellular or molecular pathways that can be used to increase drug delivery and to find ways to identify patients who may benefit from combined therapy. Targeted therapies include oncogenes, growth factors, and molecules aimed at blocking gene activity. Growth factors often govern normal cell growth. Growth factors are released from either the cell or the surrounding tissue. Cancer cells can either secrete these growth factors or respond to them, enabling them to divide out of control. A number of tumor growth factors have been identified, including those that trigger nerve tissue growth and stimulate blood vessels to grow (a process called angiogenesis). Researchers continue to identify and develop agents that can block these factors. Anti-angiogenetic compounds block blood vessel growth and the flow of nutrients and oxygen to the tumor, and may also hamper cell signaling and stop tumor cells from spreading elsewhere in the body. Clinical trials have shown that anti-angiogenetics can improve the outcome for glioblastoma multiforme tumors that recur following initial treatment with radiation and chemotherapy. More than a dozen such compounds have been identified, including bevacizumab, interferon, and endostatin. Researchers are studying the combination of anti-angiogenics and radiation or chemotherapy in treating newly diagnosed brain tumors. Other research is testing angiogenic inhibitors that have had positive results in adults on children with primary CNS tumors and in patients with tumors that haven't responded well to other tumor treatment. Oncogenes are transformed genes that are involved in cell growth and cause normal cells to divide uncontrolled and become malignant, either through mutation or over-expression. Researchers have identified specific events that lead to this mutation and are developing diagnostic and therapeutic screening tools. Certain enzymes that are involved in cell division or the copying of genetic information may be over-produced in cancer cells, causing gene mutations and uncontrolled cell growth. Several different types of kinase inhibitors—proteins that block growth-signaling enzymes without harming normal cells—have been identified and approved for cancer treatment. Scientists are testing protein-level kinase inhibitors to see of they make CNS tumors more sensitive to chemotherapy. Clinical trials for brain tumors are studying the safety and effectiveness of kinase inhibitors in children with CNS tumors and in other patients with recurring or hard to treat tumors. Anti-sense oligonucleotide technology uses short fragments of nucleic acid molecules to block gene expression in specific cells and prevent tumor growth. Unlike gene therapy, anti-sense oligonucleotide technology does not replace or substitute genetic material but inhibits the expression of select targets. This technology (also known as RNA interference, siRNA, or shRNA) may help scientists to identify additional genes involved in tumor formation and improve drug delivery with fewer side effects. Of particular interest are microRNAs (or miRNAs)—naturally occurring molecules that regulate gene expression and are involved in the formation and development of tumor stem cells. Researchers are using miRNAs to switch off tumor stem cell activity. Researchers are studying miRNAs as a possible diagnostic and therapeutic strategy for brain tumors and other forms of cancer. Biomarkers are molecules or others substances in the blood or tissue that can be used to diagnose or monitor a particular disorder, among other functions. As cells become cancerous, they can release unique proteins and other molecules into the body which scientists use to speed diagnosis and treatment. Some CNS tumor biomarkers have been found, such as the epithelial growth factor receptor (EGRF) gene. Researchers continue to search for additional clinical biomarkers of CNS tumors, to better assess risk from environmental toxins and other possible causes, and monitor and predict the outcome of CNS tumor treatment. NINDS-funded research has recently shown that mutation analysis of EGFR can be used to predict which tumors are most likely to respond to a specific class of cancer-fighting drugs. Identifying biomarkers may also lead to the development of new models of disease and novel therapies for tumor treatment. Radiation therapy research incluces testing several new anticancer drugs, either independently or in combination with other drugs. Researchers are also testing different drugs in combination with other therapies and are investigating combined therapies such as radiation and radiosurgery to effectively treat CNS tumors. Research areas being explored include tomotherapy, boron nuclear capture therapy, liquid radiation therapy, and the use of radiosensitizers. Radiosensitizers are drugs that make rapidly dividing tumor tissue more vulnerable to radiation. Early studies using radiosensitizers produced mixed results. While some trials suggested these drugs may improve survival in certain patients, other trials were found to have little benefit. Scientists are now testing an experimental drug that increases the amount of oxygen that is released from the blood into the tissues, making cancer cells more responsive to radiation and chemotherapy. Studies show the lack of oxygen in a tumor can make radiation therapy less effective. Tomotherapy combines CT scanning and intensity modulated radiation therapy to deliver small beams of high-dose radiation to the tumor while greatly reducing radiation exposure to surrounding tissue. Studies are examining the effectiveness of radiation delivery through tomotherapy and to see if long-term side effects of radiation therapy can be reduced. Boron Neutron Capture Therapy (BNCT) is an experimental treatment that uses fission to kill tumor cells. An amino acid or other drug that carries boron is injected into the patient, where it collects more readily in tumor cells than in regular tissue. A beam of low-energy neutrons is directed at the tumor from outside the body, causing the boron atoms to split (called neutron capture) and send high amounts of energy into the cancerous cells. Radiation damage to surrounding cells is extremely small. BNCT is being tested as a post-surgery treatment for patients with certain head and neck tumors. If successful, BNCT could someday be used to treat children with brain tumors. Liquid radiation therapy is an experimental treatment that uses a balloon catheter to deliver internal radiation therapy to the cavity that remains following tumor surgery. A radioactive liquid is injected into the catheter and molds the balloon to the exact edges of the cavity. The liquid stays in the balloon for several days until it and the catheter are removed. Chemotherapeutic drug research focuses on ways to better deliver drugs across the blood-brain barrier and into the site of the tumor. Since chemotherapeutic drugs work in different ways to stop tumor cells from dividing, several trials are testing whether giving more than one drug, and perhaps giving them in different ways (such as staggered delivery and low-dose, long-term treatment), may kill more tumor cells without causing or lessening damaging side effects than present therapy. Researchers are examining different levels of chemotherapeutic drugs to determine if they are less toxic to normal tissue when combined with other cancer treatments. Other studies are investigating gene therapy as a way to make cancer cells more sensitive to chemotherapy. Research areas include differentiating drugs, osmotic blood-brain barrier disruption, and convection enhanced delivery. Differentiating drugs change dividing cells into non-dividing cells and can halt tumor growth. An early study using retinoids (made from vitamin A) as an independent treatment for certain tumors showed no significant effect. Scientists are trying retinoids in combination with other chemotherapeutic drugs or treatments to slow the growth of malignant glioma. Researchers are testing different drugs and molecules to see if they can modulate the normal activity of the blood-brain barrier and better target tumor cells and associated blood vessels. Osmotic blood-brain barrier disruption uses certain drugs to open blood vessels throughout the brain. Scientists are currently studying whether certain chemotherapeutic drugs, when given with osmotic blood-brain barrier disruption, might be an effective way to kill cancer cells without harmful side effects. Convection enhanced delivery sends a continuous, uniform stream of toxic drugs into the tumor via catheters that are inserted during surgery. This drug delivery system, which bypasses the blood-brain barrier, is being evaluated in children with recurring brain cancer and in patients whose tumor location prevents its total surgical removal. Also under investigation are ways to help the body respond to improved drug delivery or other cancer treatments. Bone marrow and stem cell transplants may replace blood-forming cells that are killed by chemotherapy, radiation treatment, or a combination of therapies. These transplants, which are given after radiation or chemotherapy, help the patient’s bone marrow recover and produce healthy blood cells. Researchers are studying the effectiveness of these transplants in protecting blood cells in children with certain types of CNS tumors and in other patients with recurrent or hard to treat CNS cancer, who receive higher than normal doses of chemotherapy. Although many new approaches to treatment appear promising, it is important to remember that all potential therapies must stand the tests of well-designed, carefully controlled clinical trials and long-term follow-up of treated patients before any conclusions can be drawn about their safety or effectiveness. New trial designs are also being developed to more quickly evaluate novel agents and may involve pre-selection of patients based on the molecular abnormality in their tumor. Past research has led to improved tumor treatments and techniques for many individuals with CNS tumors. Current research promises to generate further improvements. In the years ahead, physicians and individuals who have a CNS tumor can look forward to new, more effective, less toxic forms of therapy developed through a better molecular understanding of the unique traits of CNS tumors. [1] Cellular Phone Use and Cancer Risk: Update of a Danish Nationwide Cohort. Schüz et. al., Journal of the National Cancer Institute, Vol. 98, No. 23, December 6, 2006, pp. 1703-1713. [2] Central Brain Tumor Registry of the United States, 2005-2006 [3] American Brain Tumor Foundation, 2008 [3] Central Brain Tumor Registry of the United States, 2005-2006 Where can I get more information? For
more information on neurological disorders or research programs funded
by the National Institute of Neurological Disorders and Stroke, contact
the Institute's Brain Resources and Information Network (BRAIN) at:
BRAIN Information also is available from the following organizations:
|
| Cerebral
Aneurysm What
is a cerebral aneurysm?
What causes a cerebral aneurysm? How are aneurysms classified? Who is at risk? What are the dangers? What are the symptoms? How are cerebral aneurysms diagnosed? How are cerebral aneurysms treated? Can cerebral aneurysms be prevented? What is the prognosis? What research is being done? Where can I get more information? What is a cerebral aneurysm?A cerebral aneurysm (also known as an intracranial or intracerebral aneurysm) is a weak or thin spot on a blood vessel in the brain that balloons out and fills with blood. The bulging aneurysm can put pressure on a nerve or surrounding brain tissue. It may also leak or rupture, spilling blood into the surrounding tissue (called a hemorrhage). Some cerebral aneurysms, particularly those that are very small, do not bleed or cause other problems. Cerebral aneurysms can occur anywhere in the brain, but most are located along a loop of arteries that run between the underside of the brain and the base of the skull. What causes a cerebral aneurysm?Most cerebral aneurysms are congenital, resulting from an inborn abnormality in an artery wall. Cerebral aneurysms are also more common in people with certain genetic diseases, such as connective tissue disorders and polycystic kidney disease, and certain circulatory disorders, such as arteriovenous malformations.[1] Other causes include trauma or injury to the head, high blood pressure, infection, tumors, atherosclerosis (a blood vessel disease in which fats build up on the inside of artery walls) and other diseases of the vascular system, cigarette smoking, and drug abuse. Some investigators have speculated that oral contraceptives may increase the risk of developing aneurysms. Aneurysms
that result from an infection in the arterial wall
are called mycotic aneurysms. Cancer-related
aneurysms are often associated with primary or metastatic
tumors of the head and neck. Drug abuse, particularly
the habitual use of cocaine, can inflame blood vessels
and lead to the development of brain aneurysms. [1] A congenital malformation in which a snarled tangle of arteries and veins in the brain disrupts blood flow. How are aneurysms classified?There are three types of cerebral aneurysm. A saccular aneurysm is a rounded or pouch-like sac of blood that is attached by a neck or stem to an artery or a branch of a blood vessel. Also known as a berry aneurysm (because it resembles a berry hanging from a vine), this most common form of cerebral aneurysm is typically found on arteries at the base of the brain. Saccular aneurysms occur most often in adults. A lateral aneurysm appears as a bulge on one wall of the blood vessel, while a fusiform aneurysm is formed by the widening along all walls of the vessel. Aneurysms are also classified by size. Small aneurysms are less than 11 millimeters in diameter (about the size of a standard pencil eraser), larger aneurysms are 11-25 millimeters (about the width of a dime), and giant aneurysms are greater than 25 millimeters in diameter (more than the width of a quarter). Who is at risk?Brain aneurysms can occur in anyone, at any age. They are more common in adults than in children and slightly more common in women than in men. People with certain inherited disorders are also at higher risk. All
cerebral aneurysms have the potential to rupture
and cause bleeding within the brain. The incidence
of reported ruptured aneurysm is about 10 in every
100,000 persons per year (about 27,000 patients per
year in the
What are the dangers?Aneurysms may burst and bleed into the brain, causing serious complications including hemorrhagic stroke, permanent nerve damage, or death. Once it has burst, the aneurysm may burst again and rebleed into the brain, and additional aneurysms may also occur. More commonly, rupture may cause a subarachnoid hemorrhage—bleeding into the space between the skull bone and the brain. A delayed but serious complication of subarachnoid hemorrhage is hydrocephalus, in which the excessive buildup of cerebrospinal fluid in the skull dilates fluid pathways called ventricles that can swell and press on the brain tissue. Another delayed postrupture complication is vasospasm, in which other blood vessels in the brain contract and limit blood flow to vital areas of the brain. This reduced blood flow can cause stroke or tissue damage. What are the symptoms?Most cerebral aneurysms do not show symptoms until they either become very large or burst. Small, unchanging aneurysms generally will not produce symptoms, whereas a larger aneurysm that is steadily growing may press on tissues and nerves. Symptoms may include pain above and behind the eye; numbness, weakness, or paralysis on one side of the face; dilated pupils; and vision changes. When an aneurysm hemorrhages, an individual may experience a sudden and extremely severe headache, double vision, nausea, vomiting, stiff neck, and/or loss of consciousness. Patients usually describe the headache as “the worst headache of my life” and it is generally different in severity and intensity from other headaches patients may experience. “Sentinel” or warning headaches may result from an aneurysm that leaks for days to weeks prior to rupture. Only a minority of patients have a sentinel headache prior to aneurysm rupture. Other signs that a cerebral aneurysm has burst include nausea and vomiting associated with a severe headache, a drooping eyelid, sensitivity to light, and change in mental status or level of awareness. Some individuals may have seizures. Individuals may lose consciousness briefly or go into prolonged coma. People experiencing this “worst headache,” especially when it is combined with any other symptoms, should seek immediate medical attention. How are cerebral aneurysms diagnosed?Most cerebral aneurysms go unnoticed until they rupture or are detected by brain imaging that may have been obtained for another condition. Several diagnostic methods are available to provide information about the aneurysm and the best form of treatment. The tests are usually obtained after a subarachnoid hemorrhage, to confirm the diagnosis of an aneurysm. Angiography is a dye test used to analyze the arteries or veins. An intracerebral angiogram can detect the degree of narrowing or obstruction of an artery or blood vessel in the brain, head, or neck, and can identify changes in an artery or vein such as a weak spot like an aneurysm. It is used to diagnose stroke and to precisely determine the location, size, and shape of a brain tumor, aneurysm, or blood vessel that has bled. This test is usually performed in a hospital angiography suite. Following the injection of a local anesthetic, a flexible catheter is inserted into an artery and threaded through the body to the affected artery. A small amount of contrast dye (one that is highlighted on x-rays) is released into the bloodstream and allowed to travel into the head and neck. A series of x-rays is taken and changes, if present, are noted. Computed tomography (CT) of the head is a fast, painless, noninvasive diagnostic tool that can reveal the presence of a cerebral aneurysm and determine, for those aneurysms that have burst, if blood has leaked into the brain. This is often the first diagnostic procedure ordered by a physician following suspected rupture. X-rays of the head are processed by a computer as two-dimensional cross-sectional images, or “slices,” of the brain and skull. Occasionally a contrast dye is injected into the bloodstream prior to scanning. This process, called CT angiography, produces sharper, more detailed images of blood flow in the brain arteries. CT is usually conducted at a testing facility or hospital outpatient setting. Magnetic resonance imaging (MRI) uses computer-generated radio waves and a powerful magnetic field to produce detailed images of the brain and other body structures. Magnetic resonance angiography (MRA) produces more detailed images of blood vessels. The images may be seen as either three-dimensional pictures or two-dimensional cross-slices of the brain and vessels. These painless, noninvasive procedures can show the size and shape of an unruptured aneurysm and can detect bleeding in the brain. Cerebrospinal fluid analysis may be ordered if a ruptured aneurysm is suspected. Following application of a local anesthetic, a small amount of this fluid (which protects the brain and spinal cord) is removed from the subarachnoid space—located between the spinal cord and the membranes that surround it—by surgical needle and tested to detect any bleeding or brain hemorrhage. In patients with suspected subarachnoid hemorrhage, this procedure is usually done in a hospital. How are cerebral aneurysms treated?Not all cerebral aneurysms burst. Some patients with very small aneurysms may be monitored to detect any growth or onset of symptoms and to ensure aggressive treatment of coexisting medical problems and risk factors. Each case is unique, and considerations for treating an unruptured aneurysm include the type, size, and location of the aneurysm; risk of rupture; patient’s age, health, and personal and family medical history; and risk of treatment. Two surgical options are available for treating cerebral aneurysms, both of which carry some risk to the patient (such as possible damage to other blood vessels, the potential for aneurysm recurrence and rebleeding, and the risk of post-operative stroke). Microvascular clipping involves cutting off the flow of blood to the aneurysm. Under anesthesia, a section of the skull is removed and the aneurysm is located. The neurosurgeon uses a microscope to isolate the blood vessel that feeds the aneurysm and places a small, metal, clothespin-like clip on the aneurysm’s neck, halting its blood supply. The clip remains in the patient and prevents the risk of future bleeding. The piece of the skull is then replaced and the scalp is closed. Clipping has been shown to be highly effective, depending on the location, shape, and size of the aneurysm. In general, aneurysms that are completely clipped surgically do not return. A related procedure is an occlusion, in which the surgeon clamps off (occludes) the entire artery that leads to the aneurysm. This procedure is often performed when the aneurysm has damaged the artery. An occlusion is sometimes accompanied by a bypass, in which a small blood vessel is surgically grafted to the brain artery, rerouting the flow of blood away from the section of the damaged artery. Endovascular embolization is an alternative to surgery. Once the patient has been anesthetized, the doctor inserts a hollow plastic tube (a catheter) into an artery (usually in the groin) and threads it, using angiography, through the body to the site of the aneurysm. Using a guide wire, detachable coils (spirals of platinum wire) or small latex balloons are passed through the catheter and released into the aneurysm. The coils or balloons fill the aneurysm, block it from circulation, and cause the blood to clot, which effectively destroys the aneurysm. The procedure may need to be performed more than once during the patient’s lifetime. Patients who receive treatment for aneurysm must remain in bed until the bleeding stops. Underlying conditions, such as high blood pressure, should be treated. Other treatment for cerebral aneurysm is symptomatic and may include anticonvulsants to prevent seizures and analgesics to treat headache. Vasospasm can be treated with calcium channel-blocking drugs and sedatives may be ordered if the patient is restless. A shunt may be surgically inserted into a ventricle several months following rupture if the buildup of cerebrospinal fluid is causing harmful pressure on surrounding tissue. Patients who have suffered a subarachnoid hemorrhage often need rehabilitative, speech, and occupational therapy to regain lost function and learn to cope with any permanent disability. Can cerebral aneurysms be prevented?There are no known ways to prevent a cerebral aneurysm from forming. People with a diagnosed brain aneurysm should carefully control high blood pressure, stop smoking, and avoid cocaine use or other stimulant drugs. They should also consult with a doctor about the benefits and risks of taking aspirin or other drugs that thin the blood. Women should check with their doctors about the use of oral contraceptives. What is the prognosis?An unruptured aneurysm may go unnoticed throughout a person’s lifetime. A burst aneurysm, however, may be fatal or could lead to hemorrhagic stroke, vasospasm (the leading cause of disability or death following a burst aneurysm), hydrocephalus, coma, or short-term and/or permanent brain damage. The prognosis for persons whose aneurysm has burst is largely dependent on the age and general health of the individual, other preexisting neurological conditions, location of the aneurysm, extent of bleeding (and rebleeding), and time between rupture and medical attention. It is estimated that about 40 percent of patients whose aneurysm has ruptured do not survive the first 24 hours; up to another 25 percent die from complications within 6 months. Patients who experience subarachnoid hemorrhage may have permanent neurological damage. Other individuals may recover with little or no neurological deficit. Delayed complications from a burst aneurysm may include hydrocephalus and vapospasm. Early diagnosis and treatment are important. Individuals who receive treatment for an unruptured aneurysm generally require less rehabilitative therapy and recover more quickly than persons whose aneurysm has burst. Recovery from treatment or rupture may take weeks to months. Results
of the International Subarachnoid Aneurysm Trial
(ISAT), sponsored primarily by health ministries
in the United Kingdom, France, and Canada and announced
in October 2002, found that outcome for patients
who are treated with endovascular coiling may be
superior in the short-term (1 year) to outcome for
patients whose aneurysm is treated with surgical
clipping. Long-term results of coiling procedures
are unknown and investigators need to conduct more
research on this topic, since some aneurysms can
recur after coiling. Before treatment patients
may want to consult with a specialist in both endovascular
and surgical repair of aneurysms, to help provide
greater understanding of treatment options. (The
American Association of Neurological Surgeons notes
that most of the centers involved in the ISAT were
in Europe [primarily in England], Australia, and
Canada, and that results may not be applicable to
patients in the United States,
“where practice patterns, particularly in reference
to the degree of sub-specialization of neurovascular
surgeons in major centers, are different.”[1]) [1] American Association of Neurological Surgeons/Congress of Neurological Surgeons, “Position Statement on the International Subarachnoid Aneurysm Trial (ISAT),” What research is being done?The National Institute of Neurological Disorders and Stroke (NINDS), a component of the National Institutes of Health (NIH) within the U.S. Department of Health and Human Services, is the nation’s primary supporter of research on the brain and nervous system. As part of its mission, the NINDS conducts research on intracranial aneurysms and other vascular lesions of the nervous system and supports studies through grants to medical institutions across the country. The NINDS recently sponsored the International Study of Unruptured Intracranial Aneurysms, which included more than 4,000 patients at 61 sites in the United States, Canada, and Europe. The findings suggest that the risk of rupture for most very small aneurysms (less than 7 millimeters in size) is low. The results also provide a more comprehensive look at these vascular defects and offer guidance to patients and physicians facing the difficult decision about whether or not to treat an aneurysm surgically. NINDS scientists are studying the effects of an experimental drug in treating vasospasm that occurs following rupture of a cerebral aneurysm. The drug, developed at the NIH, delivers nitric oxide to the arteries and has been shown to reverse and prevent brain artery spasms in animals. Other scientists hope to improve diagnosis and prediction of cerebral vasospasm by developing antibodies to molecules known to cause vasospasm. These molecules can be detected in the cerebrospinal fluid of subarachnoid hemorrhage patients. An additional study will compare standard treatment for subarachnoid hemorrhage to standard treatment plus transluminal balloon angioplasty immediately after severe bleeding. Transluminal balloon angioplasty involves the insertion, via catheter, of a deflated balloon through the affected artery and into the clot. The balloon is inflated to widen the artery and restore blood flow (the deflated balloon and catheter are then withdrawn). Researchers are building a new, noninvasive, high-resolution x-ray detector system that can be used to guide the placement of stents (small tube-like devices that keep blood vessels open) used to modify blood flow during treatment for brain aneurysms. Several groups of NINDS-funded researchers are conducting genetic linkage studies to identify risk factors for familial intracranial aneurysm and/or subarachnoid hemorrhage. One study hopes to establish patterns of inheritance in patients of different ethnic backgrounds. Another project is aimed at targeting and providing prevention and treatment strategies for persons who are genetically at high risk for the development of brain aneurysms. And other investigators will establish a blood and tissue sampling bank for genetic linkage and molecular analyses. Scientists are investigating the use of intraoperative hypothermia during microclip surgery as a means to improve the rate of recovery of cognitive functions and to reduce early and postoperative complications and neurological damage. Other studies are investigating ways to improve or replace the coils used in endovascular embolization. Additional research being funded by the NINDS includes the development of a new animal model of human saccular aneurysm, a new method for tissue processing that should allow routine evaluation of the biological response to implantation of occlusion devices, and a computer simulation model to evaluate the outcomes of neurosurgery in patients with cerebral aneurysms. Where can I get more information? For more information on neurological disorders or research
programs funded by the National Institute of Neurological
Disorders and Stroke, contact the Institute's Brain Resources
and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
|
|
Introduction
What is Cerebral Palsy? How Many People Have Cerebral Palsy? What Are the Early Signs? What Causes Cerebral Palsy? What are the Risk Factors? Can Cerebral Palsy Be Prevented? What Are the Different Forms? What Other Conditions Are Associated With Cerebral Palsy? How Does a Doctor Diagnose Cerebral Palsy? How is Cerebral Palsy Managed? What Specific Treatments Are Available? Drug Treatments Surgery Orthotic Devices Assistive Technology Alternative Therapies Are There Treatments for Other Conditions Associated with Cerebral Palsy? Do Adults with Cerebral Palsy Face Special Health Challenges? What Research Is Being Done? Where can I get more information? Glossary IntroductionIn the 1860s, an English surgeon named William Little wrote the first medical descriptions of a puzzling disorder that struck children in the first years of life, causing stiff, spastic muscles in their legs and, to a lesser degree, in their arms. These children had difficulty grasping objects, crawling, and walking. Unlike most other diseases that affect the brain, this condition didn’t get worse as the children grew older. Instead, their disabilities stayed relatively the same. The disorder, which was called Little's disease for many years, is now known as spastic diplegia. It is one of a group of disorders that affect the control of movement and are gathered under the umbrella term of “cerebral palsy.” Because it seemed that many of Little’s patients were born following premature or complicated deliveries, the doctor suggested their condition was the result of oxygen deprivation during birth, which damaged sensitive brain tissues controlling movement. But in 1897, the famous psychiatrist Sigmund Freud disagreed. Noting that children with cerebral palsy often had other neurological problems such as mental retardation, visual disturbances, and seizures, Freud suggested that the disorder might have roots earlier in life, during the brain's development in the womb. "Difficult birth, in certain cases," he wrote, "is merely a symptom of deeper effects that influence the development of the fetus." In spite of Freud’s observation, for many decades the belief that birth complications caused most cases of cerebral palsy was widespread among physicians, families, and even medical researchers. In the 1980s, however, scientists funded by the National Institute of Neurological Disorders and Stroke (NINDS) analyzed extensive data from more than 35,000 newborns and their mothers, and discovered that complications during birth and labor accounted for only a fraction of the infants born with cerebral palsy — probably less than 10 percent. In most cases, they could find no single, obvious cause. This finding challenged the accepted medical theory about the cause of cerebral palsy. It also stimulated researchers to search for other factors before, during, and after birth that were associated with the disorder. Advances in imaging technology, such as magnetic resonance imaging (MRI), have given researchers a way to look into the brains of infants and children with cerebral palsy and discover unique structural malformations and areas of damage. Basic science studies have identified genetic mutations and deletions associated with the abnormal development of the fetal brain. These discoveries offer provocative clues about what could be going wrong during brain development to cause the abnormalities that lead to cerebral palsy. Much of this new understanding about what causes cerebral palsy is the result of research spanning the past two decades that has been sponsored by the NINDS, the federal government’s leading supporter of neurological research. These findings from NINDS research have:
This brochure describes what cerebral palsy is, its causes, its treatments, and how it might possibly be prevented. Medical terms in italics are defined in the glossary at the back of the booklet. What is Cerebral Palsy?Doctors use the term cerebral palsy to refer to any one of a number of neurological disorders that appear in infancy or early childhood and permanently affect body movement and muscle coordination but aren’t progressive, in other words, they don’t get worse over time. The term cerebral refers to the two halves or hemispheres of the brain, in this case to the motor area of the brain’s outer layer (called the cerebral cortex), the part of the brain that directs muscle movement; palsy refers to the loss or impairment of motor function. Even though cerebral palsy affects muscle movement, it isn’t caused by problems in the muscles or nerves. It is caused by abnormalities inside the brain that disrupt the brain’s ability to control movement and posture. In some cases of cerebral palsy, the cerebral motor cortex hasn’t developed normally during fetal growth. In others, the damage is a result of injury to the brain either before, during, or after birth. In either case, the damage is not repairable and the disabilities that result are permanent. Children with cerebral palsy exhibit a wide variety of symptoms, including:
The symptoms of cerebral palsy differ in type and severity from one person to the next, and may even change in an individual over time. Some people with cerebral palsy also have other medical disorders, including mental retardation, seizures, impaired vision or hearing, and abnormal physical sensations or perceptions. Cerebral palsy doesn’t always cause profound disabilities. While one child with severe cerebral palsy might be unable to walk and need extensive, lifelong care, another with mild cerebral palsy might be only slightly awkward and require no special assistance. Cerebral palsy isn’t a disease. It isn’t contagious and it can’t be passed from one generation to the next. There is no cure for cerebral palsy, but supportive treatments, medications, and surgery can help many individuals improve their motor skills and ability to communicate with the world. How Many People Have Cerebral Palsy?The
United Cerebral Palsy (UCP) Foundation estimates that nearly
800,000 children and adults in the
Despite advances in preventing and treating certain causes of cerebral palsy, the percentage of babies who develop the condition has remained the same over the past 30 years. Improved care in neonatal intensive-care units has resulted in higher survival rates for very low birthweight babies. Many of these infants will have developmental defects in their nervous systems or suffer brain damage that will cause the characteristic symptoms of cerebral palsy. What Are the Early Signs?The
early signs of cerebral palsy usually appear before a child reaches
3 years of age. Parents are often the first to suspect
that their baby’s motor skills aren’t developing
normally. Infants with cerebral palsy frequently have developmental
delay, in which they are slow to reach developmental milestones
such as learning to roll over, sit, crawl, smile, or walk. Some
infants with cerebral palsy have abnormal muscle tone as infants.
Decreased muscle tone (hypotonia) can make them
appear relaxed, even floppy. Increased muscle tone (hypertonia) can
make them seem stiff or rigid. In some cases, an early period
of hypotonia will progress to hypertonia after the first 2 to
3 months of life. Children with cerebral palsy may also have
unusual posture or favor one side of the body when they move.
Parents who are concerned about their baby's development for any reason should contact their pediatrician. A doctor can determine the difference between a normal lag in development and a delay that could indicate cerebral palsy. What Causes Cerebral Palsy?The majority of children with cerebral palsy are born with it, although it may not be detected until months or years later. This is called congenital cerebral palsy. In the past, if doctors couldn’t identify another cause, they attributed most cases of congenital cerebral palsy to problems or complications during labor that caused asphyxia (a lack of oxygen) during birth. However, extensive research by NINDS scientists and others has shown that few babies who experience asphyxia during birth grow up to have cerebral palsy or any other neurological disorder. Birth complications, including asphyxia, are now estimated to account for only 5 to 10 percent of the babies born with congenital cerebral palsy. A small number of children have acquired cerebral palsy, which means the disorder begins after birth. In these cases, doctors can often pinpoint a specific reason for the problem, such as brain damage in the first few months or years of life, brain infections such as bacterial meningitis or viral encephalitis, or head injury from a motor vehicle accident, a fall, or child abuse. What causes the remaining 90 to 95 percent? Research has given us a bigger and more accurate picture of the kinds of events that can happen during early fetal development, or just before, during, or after birth, that cause the particular types of brain damage that will result in congenital cerebral palsy. There are multiple reasons why cerebral palsy happens – as the result of genetic abnormalities, maternal infections or fevers, or fetal injury, for example. But in all cases the disorder is the result of four types of brain damage that cause its characteristic symptoms: Damage to the white matter of the brain (periventricular leukomalacia [PVL]). The white matter of the brain is responsible for transmitting signals inside the brain and to the rest of the body. PVL describes a type of damage that looks like tiny holes in the white matter of an infant’s brain. These gaps in brain tissue interfere with the normal transmission of signals. There are a number of events that can cause PVL, including maternal or fetal infection. Researchers have also identified a period of selective vulnerability in the developing fetal brain, a period of time between 26 and 34 weeks of gestation, in which periventricular white matter is particularly sensitive to insults and injury. Abnormal development of the brain (cerebral dysgenesis). Any interruption of the normal process of brain growth during fetal development can cause brain malformations that interfere with the transmission of brain signals. The fetal brain is particularly vulnerable during the first 20 weeks of development. Mutations in the genes that control brain development during this early period can keep the brain from developing normally. Infections, fevers, trauma, or other conditions that cause unhealthy conditions in the womb also put an unborn baby’s nervous system at risk. Bleeding in the brain (intracranial hemorrhage). Intracranial hemorrhage describes bleeding inside the brain caused by blocked or broken blood vessels. A common cause of this kind of damage is fetal stroke. Some babies suffer a stroke while still in the womb because of blood clots in the placenta that block blood flow. Other types of fetal stroke are caused by malformed or weak blood vessels in the brain or by blood-clotting abnormalities. Maternal high blood pressure (hypertension) is a common medical disorder during pregnancy that has been known to cause fetal stroke. Maternal infection, especially pelvic inflammatory disease, has also been shown to increase the risk of fetal stroke. Brain damage caused by a lack of oxygen in the brain (hypoxic-ischemic encephalopathy or intrapartum asphyxia). Asphyxia, a lack of oxygen in the brain caused by an interruption in breathing or poor oxygen supply, is common in babies due to the stress of labor and delivery. But even though a newborn’s blood is equipped to compensate for short-term low levels of oxygen, if the supply of oxygen is cut off or reduced for lengthy periods, an infant can develop a type of brain damage called hypoxic-ischemic encephalopathy, which destroys tissue in the cerebral motor cortex and other areas of the brain. This kind of damage can also be caused by severe maternal low blood pressure, rupture of the uterus, detachment of the placenta, or problems involving the umbilical cord. What are the Risk Factors?Just as there are particular types of brain damage that cause cerebral palsy, there are also certain medical conditions or events that can happen during pregnancy and delivery that will increase a baby’s risk of being born with cerebral palsy. Research scientists have examined thousands of expectant mothers, followed them through childbirth, and monitored their children’s early neurological development to establish these risk factors. If a mother or her baby has any of these risk factors, it doesn’t mean that cerebral palsy is inevitable, but it does increase the chance for the kinds of brain damage that cause it. Low birthweight and premature birth. The risk of cerebral palsy is higher among babies who weigh less than 5 ½ pounds at birth or are born less than 37 weeks into pregnancy. The risk increases as birthweight falls or weeks of gestation shorten. Intensive care for premature infants has improved dramatically over the course of the past 30 years. Babies born extremely early are surviving, but with medical problems that can put them at risk for cerebral palsy. Although normal- or heavier-weight babies are at relatively low individual risk for cerebral palsy, term or near-term babies still make up half of the infants born with the condition. Multiple births. Twins, triplets, and other multiple births -- even those born at term -- are linked to an increased risk of cerebral palsy. The death of a baby’s twin or triplet further increases the risk. Infections during pregnancy. Infectious diseases caused by viruses, such as toxoplasmosis, rubella (German measles), cytomegalovirus, and herpes, can infect the womb and placenta. Researchers currently think that maternal infection leads to elevated levels of immune system cells called cytokines that circulate in the brain and blood of the fetus. Cytokines respond to infection by triggering inflammation. Inflammation may then go on to cause central nervous system damage in an unborn baby. Maternal fever during pregnancy or delivery can also set off this kind of inflammatory response. Blood
type incompatibility. Rh incompatibility is
a condition that develops when a mother’s Rh blood
type (either positive or negative) is different from the
blood type of her baby. Because blood cells from
the baby and mother mix during pregnancy, if a mother is
negative and her baby positive, for example, the mother’s
system won’t tolerate the presence of Rh-positive
red blood cells. Her body will begin to make antibodies
that will attack and kill her baby’s blood cells. Rh
incompatibility is routinely tested for and treated in
the
Exposure to toxic substances. Mothers who have been exposed to toxic substances during pregnancy, such as methyl mercury, are at a heightened risk of having a baby with cerebral palsy. Mothers with thyroid abnormalities, mental retardation, or seizures. Mothers with any of these conditions are slightly more likely to have a child with cerebral palsy. There are also medical conditions during labor and delivery, and immediately after delivery, that act as warning signs for an increased risk of cerebral palsy. Knowing these warning signs helps doctors keep a close eye on children who face a higher risk. However, parents shouldn’t become too alarmed if their baby has one or more of these conditions at birth. Most of these children will not develop cerebral palsy. Warning signs include: Breech presentation. Babies with cerebral palsy are more likely to be in a breech position (feet first) instead of head first at the beginning of labor. Complicated labor and delivery. A baby who has vascular or respiratory problems during labor and delivery may already have suffered brain damage or abnormalities. Small for gestational age. Babies born smaller than normal for their gestational age are at risk for cerebral palsy because of factors that kept them from growing naturally in the womb. Low Apgar score. The Apgar score is a numbered rating that reflects a newborn's condition. To determine an Apgar score, doctors periodically check a baby's heart rate, breathing, muscle tone, reflexes, and skin color during the first minutes after birth. They then assign points; the higher the score, the more normal a baby's condition. A low score at 10-20 minutes after delivery is often considered an important sign of potential problems such as cerebral palsy. Jaundice. More than 50 percent of newborns develop jaundice after birth when bilirubin, a substance normally found in bile, builds up faster than their livers can break it down and pass it from the body. Severe, untreated jaundice can cause a neurological condition known as kernicterus, which kills brain cells and can cause deafness and cerebral palsy. Seizures. An infant who has seizures faces a higher risk of being diagnosed later in childhood with cerebral palsy. Can Cerebral Palsy Be Prevented?Cerebral palsy related to genetic abnormalities is not preventable, but a few of the risk factors for congenital cerebral palsy can be managed or avoided. For example, rubella, or German measles, is preventable if women are vaccinated against the disease before becoming pregnant. Rh incompatibilities can also be managed early in pregnancy. But there are still risk factors that can’t be controlled or avoided in spite of medical intervention. For example, the use of electronic fetal monitoring machines to keep track of an unborn baby’s heartbeat during labor, and the use of emergency cesarean section surgery when there are significant signs of fetal distress, haven’t lowered the numbers of babies born with cerebral palsy. Interventions to treat other prenatal causes of cerebral palsy, such as therapies to prevent prenatal stroke or antibiotics to cure intrauterine infections, are either difficult to administer or haven’t yet been proven to lower the risk of cerebral palsy in vulnerable infants. Fortunately, acquired cerebral palsy, often due to head injury, is preventable using common safety tactics, such as using car seats for infants and toddlers, and making sure young children wear helmets when they ride bicycles. In addition, common sense measures around the household, such as supervising babies and young children closely when they bathe, can reduce the risk of accidental injury. Despite the best efforts of parents and physicians, however, children will still be born with cerebral palsy. Since in many cases the cause or causes of cerebral palsy aren’t fully known, little can currently be done to prevent it. As investigators learn more about the causes of cerebral palsy through basic and clinical research, doctors and parents will know more about how to prevent this disorder. What Are the Different Forms?The specific forms of cerebral palsy are determined by the extent, type, and location of a child’s abnormalities. Doctors classify cerebral palsy according to the type of movement disorder involved -- spastic (stiff muscles), athetoid (writhing movements), or ataxic (poor balance and coordination) -- plus any additional symptoms. Doctors will often describe the type of cerebral palsy a child has based on which limbs are affected. The names of the most common forms of cerebral palsy use Latin terms to describe the location or number of affected limbs, combined with the words for weakened (paresis) or paralyzed (plegia). For example, hemiparesis (hemi = half) indicates that only one side of the body is weakened. Quadriplegia (quad = four) means all four limbs are paralyzed. Spastic hemiplegia/hemiparesis. This type of cerebral palsy typically affects the arm and hand on one side of the body, but it can also include the leg. Children with spastic hemiplegia generally walk later and on tip-toe because of tight heel tendons. The arm and leg of the affected side are frequently shorter and thinner. Some children will develop an abnormal curvature of the spine (scoliosis). Depending on the location of the brain damage, a child with spastic hemiplegia may also have seizures. Speech will be delayed and, at best, may be competent, but intelligence is usually normal. Spastic diplegia/diparesis. In this type of cerebral palsy, muscle stiffness is predominantly in the legs and less severely affects the arms and face, although the hands may be clumsy. Tendon reflexes are hyperactive. Toes point up. Tightness in certain leg muscles makes the legs move like the arms of a scissor. Children with this kind of cerebral palsy may require a walker or leg braces. Intelligence and language skills are usually normal. Spastic quadriplegia/quadriparesis. This is the most severe form of cerebral palsy, often associated with moderate-to-severe mental retardation. It is caused by widespread damage to the brain or significant brain malformations. Children will often have severe stiffness in their limbs but a floppy neck. They are rarely able to walk. Speaking and being understood are difficult. Seizures can be frequent and hard to control. Dyskinetic cerebral palsy (also includes athetoid, choreoathetoid, and dystonic cerebral palsies). This type of cerebral palsy is characterized by slow and uncontrollable writhing movements of the hands, feet, arms, or legs. In some children, hyperactivity in the muscles of the face and tongue makes them grimace or drool. They find it difficult to sit straight or walk. Children may also have problems coordinating the muscle movements required for speaking. Intelligence is rarely affected in these forms of cerebral palsy. Ataxic cerebral palsy. This rare type of cerebral palsy affects balance and depth perception. Children will often have poor coordination and walk unsteadily with a wide-based gait, placing their feet unusually far apart. They have difficulty with quick or precise movements, such as writing or buttoning a shirt. They may also have intention tremor, in which a voluntary movement, such as reaching for a book, is accompanied by trembling that gets worse the closer their hand gets to the object. Mixed types. It is common for children to have symptoms that don’t correspond to any single type of cerebral palsy. Their symptoms are a mix of types. For example, a child with mixed cerebral palsy may have some muscles that are too tight and others that are too relaxed, creating a mix of stiffness and floppiness. What Other Conditions Are Associated With Cerebral Palsy?Many individuals with cerebral palsy have no additional medical disorders. However, because cerebral palsy involves the brain and the brain controls so many of the body’s functions, cerebral palsy can also cause seizures, impair intellectual development, and affect vision, hearing, and behavior. Coping with these disabilities may be even more of a challenge than coping with the motor impairments of cerebral palsy. These additional medical conditions include: Mental retardation. Two-thirds of individuals with cerebral palsy will be intellectually impaired. Mental impairment is more common among those with spastic quadriplegia than in those with other types of cerebral palsy, and children who have epilepsy and an abnormal electroencephalogram (EEG) or MRI are also more likely to have mental retardation. Seizure disorder. As many as half of all children with cerebral palsy have seizures. Seizures can take the form of the classic convulsions of tonic-clonic seizures or the less obvious focal (partial) seizures, in which the only symptoms may be muscle twitches or mental confusion. Delayed growth and development. A syndrome called failure to thrive is common in children with moderate-to-severe cerebral palsy, especially those with spastic quadriparesis. Failure to thrive is a general term doctors use to describe children who lag behind in growth and development. In babies this lag usually takes the form of too little weight gain. In young children it can appear as abnormal shortness, and in teenagers it may appear as a combination of shortness and lack of sexual development. In addition, the muscles and limbs affected by cerebral palsy tend to be smaller than normal. This is especially noticeable in children with spastic hemiplegia because limbs on the affected side of the body may not grow as quickly or as long as those on the normal side. Spinal deformities. Deformities of the spine -- curvature (scoliosis), humpback (kyphosis), and saddle back (lordosis) -- are associated with cerebral palsy. Spinal deformities can make sitting, standing, and walking difficult and cause chronic back pain. Impaired vision, hearing, or speech. A large number of children with cerebral palsy have strabismus, commonly called “cross eyes,” in which the eyes are misaligned because of differences between the left and right eye muscles. In an adult, strabismus causes double vision. In children, the brain adapts to the condition by ignoring signals from one of the misaligned eyes. Untreated, this can lead to poor vision in one eye and can interfere with the ability to judge distance. In some cases, doctors will recommend surgery to realign the muscles. Children with hemiparesis may have hemianopia, which is defective vision or blindness that blurs the normal field of vision in one eye. In homonymous hemianopia, the impairment affects the same part of the visual field in both eyes. Impaired hearing is also more frequent among those with cerebral palsy than in the general population. Speech and language disorders, such as difficulty forming words and speaking clearly, are present in more than a third of those with cerebral palsy. Drooling. Some individuals with cerebral palsy drool because they have poor control of the muscles of the throat, mouth, and tongue. Drooling can cause severe skin irritation. Because it is socially unacceptable, drooling may also isolate children from their peers. Incontinence. A common complication of cerebral palsy is incontinence, caused by poor control of the muscles that keep the bladder closed. Incontinence can take the form of bed-wetting, uncontrolled urination during physical activities, or slow leaking of urine throughout the day. Abnormal sensations and perceptions. Some children with cerebral palsy have difficulty feeling simple sensations, such as touch. They may have stereognosia, which makes it difficult to perceive and identify objects using only the sense of touch. A child with stereognosia, for example, would have trouble closing his eyes and sensing the difference between a hard ball or a sponge ball placed in his hand. How Does a Doctor Diagnose Cerebral Palsy?Early signs of cerebral palsy may be present from birth. Most children with cerebral palsy are diagnosed during the first 2 years of life. But if a child’s symptoms are mild, it can be difficult for a doctor to make a reliable diagnosis before the age of 4 or 5. Nevertheless, if a doctor suspects cerebral palsy, he or she will most likely schedule an appointment to observe the child and talk to the parents about their child’s physical and behavioral development. Doctors diagnose cerebral palsy by evaluating a child’s motor skills and taking a careful and thorough look at their medical history. In addition to checking for the most characteristic symptoms -- slow development, abnormal muscle tone, and unusual posture -- a doctor also has to rule out other disorders that could cause similar symptoms. Most important, a doctor has to determine that the child's condition is not getting worse. Although symptoms may change over time, cerebral palsy by definition is not progressive. If a child is continuously losing motor skills, the problem more likely begins elsewhere – such as a genetic or muscle disease, metabolism disorder, or tumors in the nervous system. A comprehensive medical history, special diagnostic tests, and, in some cases, repeated check-ups can help confirm that other disorders are not at fault. Additional tests are often used to rule out other movement disorders that could cause the same symptoms as cerebral palsy. Neuroimaging techniques that allow doctors to look into the brain (such as an MRI scan) can detect abnormalities that indicate a potentially treatable movement disorder. If it is cerebral palsy, an MRI scan can also show a doctor the location and type of brain damage. Neuroimaging methods include:
On rare occasions, metabolic disorders can masquerade as cerebral palsy and some children will require additional tests to rule them out. Most of the childhood metabolic disorders have characteristic brain abnormalities or malformations that will show up in an MRI. Other types of disorders can also be mistaken for cerebral palsy. For example, coagulation disorders (which prevent blood from clotting) can cause prenatal or perinatal strokes that damage the brain and cause symptoms characteristic of cerebral palsy. Because stroke is so often the cause of hemiplegic cerebral palsy, a doctor may find it necessary to perform diagnostic testing on children with this kind of cerebral palsy to rule out the presence of a coagulation disorder. If left undiagnosed, coagulation disorders can cause additional strokes and more extensive brain damage. To confirm a diagnosis of cerebral palsy, a doctor may refer a child to additional doctors with specialized knowledge and training, such as a child neurologist, developmental pediatrician, ophthalmologist (eye doctor), or otologist (ear doctor). Additional observations help a doctor make a more accurate diagnosis and begin to develop a specific plan for treatment. How is Cerebral Palsy Managed?Cerebral palsy can’t be cured, but treatment will often improve a child's capabilities. Many children go on to enjoy near-normal adult lives if their disabilities are properly managed. In general, the earlier treatment begins, the better chance children have of overcoming developmental disabilities or learning new ways to accomplish the tasks that challenge them. There is no standard therapy that works for every individual with cerebral palsy. Once the diagnosis is made, and the type of cerebral palsy is determined, a team of health care professionals will work with a child and his or her parents to identify specific impairments and needs, and then develop an appropriate plan to tackle the core disabilities that affect the child’s quality of life. A comprehensive management plan will pull in a combination of health professionals with expertise in the following: physical therapy to improve walking and gait, stretch spastic muscles, and prevent deformities; occupational therapy to develop compensating tactics for everyday activities such as dressing, going to school, and participating in day-to-day activities; speech therapy to address swallowing disorders, speech impediments, and other obstacles to communication; counseling and behavioral therapy to address emotional and psychological needs and help children cope emotionally with their disabilities; drugs to control seizures, relax muscle spasms, and alleviate pain; surgery to correct anatomical abnormalities or release tight muscles; braces and other orthotic devices to compensate for muscle imbalance, improve posture and walking, and increase independent mobility; mechanical aids such as wheelchairs and rolling walkers for individuals who are not independently mobile; and communication aids such as computers, voice synthesizers, or symbol boards to allow severely impaired individuals to communicate with others. Doctors use tests and evaluation scales to determine a child’s level of disability, and then make decisions about the types of treatments and the best timing and strategy for interventions. Early intervention programs typically provide all the required therapies within a single treatment center. Centers also focus on parents’ needs, often offering support groups, babysitting services, and respite care. The members of the treatment team for a child with cerebral palsy will most likely include the following: A physician, such as a pediatrician, pediatric neurologist, or pediatric physiatrist, who is trained to help developmentally disabled children. This doctor, who often acts as the leader of the treatment team, integrates the professional advice of all team members into a comprehensive treatment plan, makes sure the plan is implemented properly, and follows the child’s progress over a number of years. An orthopedist, a surgeon who specializes in treating the bones, muscles, tendons, and other parts of the skeletal system. An orthopedist is often brought in to diagnose and treat muscle problems associated with cerebral palsy. A physical therapist, who designs and puts into practice special exercise programs to improve strength and functional mobility. An occupational therapist, who teaches the skills necessary for day-to-day living, school, and work. A speech and language pathologist, who specializes in diagnosing and treating disabilities relating to difficulties with swallowing and communication. A social worker, who helps individuals and their families locate community assistance and education programs. A psychologist, who helps individuals and their families cope with the special stresses and demands of cerebral palsy. In some cases, psychologists may also oversee therapy to modify unhelpful or destructive behaviors. An educator, who may play an especially important role when mental retardation or learning disabilities present a challenge to education. Regardless of age or the types of therapy that are used, treatment doesn’t end when an individual with cerebral palsy leaves the treatment center. Most of the work is done at home. Members of the treatment team often act as coaches, giving parents and children techniques and strategies to practice at home. Studies have shown that family support and personal determination are two of the most important factors in helping individuals with cerebral palsy reach their long-term goals. While mastering specific skills is an important focus of treatment on a day-to-day basis, the ultimate goal is to help children grow into adulthood with as much independence as possible. As a child with cerebral palsy grows older, the need for therapy and the kinds of therapies required, as well as support services, will likely change. Counseling for emotional and psychological challenges may be needed at any age, but is often most critical during adolescence. Depending on their physical and intellectual abilities, adults may need help finding attendants to care for them, a place to live, a job, and a way to get to their place of employment. Addressing the needs of parents and caregivers is also an important component of the treatment plan. The well-being of an individual with cerebral palsy depends upon the strength and well-being of his or her family. For parents to accept a child’s disabilities and come to grips with the extent of their caregiving responsibilities will take time and support from health care professionals. Family-centered programs in hospitals and clinics and community-based organizations usually work together with families to help them make well-informed decisions about the services they need. They also coordinate services to get the most out of treatment. A good program will encourage the open exchange of information, offer respectful and supportive care, encourage partnerships between parents and the health care professionals they work with, and acknowledge that although medical specialists may be the experts, it’s parents who know their children best. What Specific Treatments Are Available?Physical therapy, usually begun in the first few years of life or soon after the diagnosis is made, is a cornerstone of cerebral palsy treatment. Physical therapy programs use specific sets of exercises and activities to work toward two important goals: preventing weakening or deterioration in the muscles that aren’t being used (disuse atrophy), and keeping muscles from becoming fixed in a rigid, abnormal position (contracture). Resistive exercise programs (also called strength training) and other types of exercise are often used to increase muscle performance, especially in children and adolescents with mild cerebral palsy. Daily bouts of exercise keep muscles that aren’t normally used moving and active and less prone to wasting away. Exercise also reduces the risk of contracture, one of the most common and serious complications of cerebral palsy. Normally growing children stretch their muscles and tendons as they run, walk, and move through their daily activities. This insures that their muscles grow at the same rate as their bones. But in children with cerebral palsy, spasticity prevents muscles from stretching. As a result, their muscles don’t grow fast enough to keep up with their lengthening bones. The muscle contracture that results can set back the gains in function they’ve made. Physical therapy alone or in combination with special braces (called orthotic devices) helps prevent contracture by stretching spastic muscles. Occupational therapy. This kind of therapy focuses on optimizing upper body function, improving posture, and making the most of a child’s mobility. An occupational therapist helps a child master the basic activities of daily living, such as eating, dressing, and using the bathroom alone. Fostering this kind of independence boosts self-reliance and self-esteem, and also helps reduce demands on parents and caregivers. Recreational therapies. Recreational therapies, such as therapeutic horseback riding (also called hippotherapy), are sometimes used with mildly impaired children to improve gross motor skills. Parents of children who participate in recreational therapies usually notice an improvement in their child’s speech, self-esteem, and emotional well-being. Controversial
physical therapies. "Patterning" is a physical
therapy based on the principle that children with cerebral
palsy should be taught motor skills in the same sequence
in which they develop in normal children. In this
controversial approach, the therapist begins by teaching
a child elementary movements such as crawling -- regardless
of age – before moving on to walking skills. Some
experts and organizations, including the
Experts
have similar reservations about the Bobath technique (which
is also called “neurodevelopmental treatment”),
named for a husband and wife team who pioneered the approach
in
The
Bobath technique has had a widespread influence on the core
physical therapies of cerebral palsy treatment, but there
is no evidence that the technique improves motor control. The
Conductive
education, developed in
Speech and language therapy. About 20 percent of children with cerebral palsy are unable to produce intelligible speech. They also experience challenges in other areas of communication, such as hand gestures and facial expressions, and they have difficulty participating in the basic give and take of a normal conversation. These challenges will last throughout their lives. Speech and language therapists (also known as speech therapists or speech-language pathologists) observe, diagnose, and treat the communication disorders associated with cerebral palsy. They use a program of exercises to teach children how to overcome specific communication difficulties. For example, if a child has difficulty saying words that begin with "b," the therapist may suggest daily practice with a list of "b" words, increasing their difficulty as each list is mastered. Other kinds of exercises help children master the social skills involved in communicating by teaching them to keep their head up, maintain eye contact, and repeat themselves when they are misunderstood. Speech therapists can also help children with severe disabilities learn how to use special communication devices, such as a computer with a voice synthesizer, or a special board covered with symbols of everyday objects and activities to which a child can point to indicate his or her wishes. Speech interventions often use a child’s family members and friends to reinforce the lessons learned in a therapeutic setting. This kind of indirect therapy encourages people who are in close daily contact with a child to create opportunities for him or her to use their new skills in conversation. Treatments for problems with eating and drooling are often necessary when children with cerebral palsy have difficulty eating and drinking because they have little control over the muscles that move their mouth, jaw, and tongue. They are also at risk for breathing food or fluid into the lungs. Some children develop gastroesophageal reflux disease (GERD, commonly called heartburn) in which a weak diaphragm can’t keep stomach acids from spilling into the esophagus. The irritation of the acid can cause bleeding and pain. Individuals with cerebral palsy are also at risk for malnutrition, recurrent lung infections, and progressive lung disease. The individuals most at risk for these problems are those with spastic quadriplegia. Initially, children should be evaluated for their swallowing ability, which is usually done with a modified barium swallow study. Recommendations regarding diet modifications will be derived from the results of this study. In severe cases where swallowing problems are causing malnutrition, a doctor may recommend tube feeding, in which a tube delivers food and nutrients down the throat and into the stomach, or gastrostomy, in which a surgical opening allows a tube to be placed directly into the stomach. Although numerous treatments for drooling have been tested over the years, there is no one treatment that helps reliably. Anticholinergic drugs – such as glycopyrolate -- can reduce the flow of saliva but may cause unpleasant side effects, such as dry mouth, constipation, and urinary retention. Surgery, while sometimes effective, carries the risk of complications. Some children benefit from biofeedback techniques that help them recognize more quickly when their mouths fall open and they begin to drool. Intraoral devices (devices that fit into the mouth) that encourage better tongue positioning and swallowing are still being evaluated, but appear to reduce drooling for some children. Drug TreatmentsOral medications such as diazepam, baclofen, dantrolene sodium, and tizanidine are usually used as the first line of treatment to relax stiff, contracted, or overactive muscles. These drugs are easy to use, except that dosages high enough to be effective often have side effects, among them drowsiness, upset stomach, high blood pressure, and possible liver damage with long-term use. Oral medications are most appropriate for children who need only mild reduction in muscle tone or who have widespread spasticity. Doctors also sometimes use alcohol “washes” -- injections of alcohol into muscles -- to reduce spasticity. The benefits last from a few months to 2 years or more, but the adverse effects include a significant risk of pain or numbness, and the procedure requires a high degree of skill to target the nerve. The availability of new and more precise methods to deliver antispasmodic medications is moving treatment for spasticity toward chemodenervation, in which injected drugs are used to target and relax muscles. Botulinum toxin (BT-A), injected locally, has become a standard treatment for overactive muscles in children with spastic movement disorders such as cerebral palsy. BT-A relaxes contracted muscles by keeping nerve cells from over-activating muscle. Although BT-A is not approved by the Food and Drug Administration (FDA) for treating cerebral palsy, since the 1990s doctors have been using it off-label to relax spastic muscles. A number of studies have shown that it reduces spasticity and increases the range of motion of the muscles it targets. The relaxing effect of a BT-A injection lasts approximately 3 months. Undesirable side effects are mild and short-lived, consisting of pain upon injection and occasionally mild flu-like symptoms. BT-A injections are most effective when followed by a stretching program including physical therapy and splinting. BT-A injections work best for children who have some control over their motor movements and have a limited number of muscles to treat, none of which is fixed or rigid. Because BT-A does not have FDA approval to treat spasticity in children, parents and caregivers should make sure that the doctor giving the injection is trained in the procedure and has experience using it in children. Intrathecal baclofen therapy uses an implantable pump to deliver baclofen, a muscle relaxant, into the fluid surrounding the spinal cord. Baclofen works by decreasing the excitability of nerve cells in the spinal cord, which then reduces muscle spasticity throughout the body. Because it is delivered directly into the nervous system, the intrathecal dose of baclofen can be as low as one one-hundredth of the oral dose. Studies have shown it reduces spasticity and pain and improves sleep. The pump is the size of a hockey puck and is implanted in the abdomen. It contains a refillable reservoir connected to an alarm that beeps when the reservoir is low. The pump is programmable with an electronic telemetry wand. The program can be adjusted if muscle tone is worse at certain times of the day or night. The baclofen pump carries a small but significant risk of serious complications if it fails or is programmed incorrectly, if the catheter becomes twisted or kinked, or if the insertion site becomes infected. Undesirable, but infrequent, side effects include overrelaxation of the muscles, sleepiness, headache, nausea, vomiting, dizziness, and constipation. As a muscle-relaxing therapy, the baclofen pump is most appropriate for individuals with chronic, severe stiffness or uncontrolled muscle movement throughout the body. Doctors have successfully implanted the pump in children as young as 3 years of age. SurgeryOrthopedic surgery is often recommended when spasticity and stiffness are severe enough to make walking and moving about difficult or painful. For many people with cerebral palsy, improving the appearance of how they walk – their gait – is also important. A more upright gait with smoother transitions and foot placements is the primary goal for many children and young adults. In the operating room, surgeons can lengthen muscles and tendons that are proportionately too short. But first, they have to determine the specific muscles responsible for the gait abnormalities. Finding these muscles can be difficult. It takes more than 30 major muscles working at the right time using the right amount of force to walk two strides with a normal gait. A problem with any of those muscles can cause an abnormal gait. In addition, because the body makes natural adjustments to compensate for muscle imbalances, these adjustments could appear to be the problem, instead of a compensation. In the past, doctors relied on clinical examination, observation of the gait, and the measurement of motion and spasticity to determine the muscles involved. Now, doctors have a diagnostic technique known as gait analysis. Gait analysis uses cameras that record how an individual walks, force plates that detect when and where feet touch the ground, a special recording technique that detects muscle activity (known as electromyography), and a computer program that gathers and analyzes the data to identify the problem muscles. Using gait analysis, doctors can precisely locate which muscles would benefit from surgery and how much improvement in gait can be expected. The timing of orthopedic surgery has also changed in recent years. Previously, orthopedic surgeons preferred to perform all of the necessary surgeries a child needed at the same time, usually between the ages of 7 and 10. Because of the length of time spent in recovery, which was generally several months, doing them all at once shortened the amount of time a child spent in bed. Now most of the surgical procedures can be done on an outpatient basis or with a short inpatient stay. Children usually return to their normal lifestyle within a week. Consequently, doctors think it is much better to stagger surgeries and perform them at times appropriate to a child’s age and level of motor development. For example, spasticity in the upper leg muscles (the adductors), which causes a “scissor pattern” walk, is a major obstacle to normal gait. The optimal age to correct this spasticity with adduction release surgery is 2 to 4 years of age. On the other hand, the best time to perform surgery to lengthen the hamstrings or Achilles tendon is 7 to 8 years of age. If adduction release surgery is delayed so that it can be performed at the same time as hamstring lengthening, the child will have learned to compensate for spasticity in the adductors. By the time the hamstring surgery is performed, the child’s abnormal gait pattern could be so ingrained that it might not be easily corrected. With shorter recovery times and new, less invasive surgical techniques, doctors can schedule surgeries at times that take advantage of a child’s age and developmental abilities for the best possible result. Selective dorsal rhizotomy (SDR) is a surgical procedure recommended only for cases of severe spasticity when all of the more conservative treatments – physical therapy, oral medications, and intrathecal baclofen -- have failed to reduce spasticity or chronic pain. In the procedure, a surgeon locates and selectively severs overactivated nerves at the base of the spinal column. Because it reduces the amount of stimulation that reaches muscles via the nerves, SDR is most commonly used to relax muscles and decrease chronic pain in one or both of the lower or upper limbs. It is also sometimes used to correct an overactive bladder. Potential side effects include sensory loss, numbness, or uncomfortable sensations in limb areas once supplied by the severed nerve. Even though the use of microsurgery techniques has refined the practice of SDR surgery, there is still controversy about how selective SDR actually is. Some doctors have concerns since it is invasive and irreversible and may only achieve small improvements in function. Although recent research has shown that combining SDR with physical therapy reduces spasticity in some children, particularly those with spastic diplegia, whether or not it improves gait or function has still not been proven. Ongoing research continues to look at this surgery's effectiveness. Spinal cord stimulation was developed in the 1980s to treat spinal cord injury and other neurological conditions involving motor neurons. An implanted electrode selectively stimulates nerves at the base of the spinal cord to inhibit and decrease nerve activity. The effectiveness of spinal cord stimulation for the treatment of cerebral palsy has yet to be proven in clinical studies. It is considered a treatment alternative only when other conservative or surgical treatments have been unsuccessful at relaxing muscles or relieving pain. Orthotic DevicesOrthotic devices – such as braces and splints – use external force to correct muscle abnormalities. The technology of orthotics has advanced over the past 30 years from metal rods that hooked up to bulky orthopedic shoes, to appliances that are individually molded from high-temperature plastics for a precise fit. Ankle-foot orthoses are frequently prescribed for children with spastic diplegia to prevent muscle contracture and to improve gait. Splints are also used to correct spasticity in the hand muscles. Assistive TechnologyDevices that help individuals move about more easily and communicate successfully at home, at school, or in the workplace can help a child or adult with cerebral palsy overcome physical and communication limitations. There are a number of devices that help individuals stand straight and walk, such as postural support or seating systems, open-front walkers, quadrupedal canes (lightweight metal canes with four feet), and gait poles. Electric wheelchairs let more severely impaired adults and children move about successfully. The computer is probably the most dramatic example of a communication device that can make a big difference in the lives of people with cerebral palsy. Equipped with a computer and voice synthesizer, a child or adult with cerebral palsy can communicate successfully with others. For example, a child who is unable to speak or write but can make head movements may be able to control a computer using a special light pointer that attaches to a headband. Alternative TherapiesTherapeutic (subthreshold) electrical stimulation, also called neuromuscular electrical stimulation (NES), pulses electricity into the motor nerves to stimulate contraction in selective muscle groups. Many studies have demonstrated that NES appears to increase range of motion and muscular strength. Threshold electrical stimulation, which involves the application of electrical stimulation at an intensity too low to stimulate muscle contraction, is a controversial therapy. Studies have not been able to demonstrate its effectiveness or any significant improvement with its use. Hyperbaric oxygen therapy. Some children have cerebral palsy as the result of brain damage from oxygen deprivation. Proponents of hyperbaric oxygen therapy propose that the brain tissue surrounding the damaged area can be “awakened” by forcing high concentrations of oxygen into the body under greater than atmospheric pressure. A recent study compared a group of children who received no hyperbaric treatment to a group that received 40 treatments over 8 weeks. On every measure of function (gross motor, cognitive, communication, and memory) at the end of 2 months of treatment and after a further 3 months of follow up, the two groups were identical in outcome. There was no added benefit from hyperbaric oxygen therapy. Are There Treatments for Other Conditions Associated with Cerebral Palsy?Epilepsy. Twenty to 40 percent of children with mental retardation and cerebral palsy also have epilepsy. Doctors usually prescribe medications to control seizures. The classic medications for this purpose are phenobarbital, phenytoin, carbamazepine, and valproate. Although these drugs generally are effective in controlling seizures, their use is hampered by harmful or unpleasant side effects. Treatment for epilepsy has advanced significantly with the development of new medications that have fewer side effects. These drugs include felbamate, gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, vigabatrin, and zonisamide. In general, drugs are prescribed based on the type of seizures an individual experiences, since no one drug controls all types. Some individuals may need a combination of two or more drugs to achieve good seizure control. Incontinence. Medical treatments for incontinence include special exercises, biofeedback, prescription drugs, surgery, or surgically implanted devices to replace or aid muscles. Specially designed absorbent undergarments can also be used to protect against accidental leaks. Osteopenia. Children with cerebral palsy who aren’t able to walk risk developing poor bone density (osteopenia), which makes them more likely to break bones. In a study of older Americans funded by the National Institutes of Health (NIH), a family of drugs called bisphosphonates, which was recently approved by the FDA to treat mineral loss in elderly patients, also appeared to increase bone mineral density. Doctors may choose to selectively prescribe the drug off-label to children to prevent osteopenia. Pain. Pain can be a problem for people with cerebral palsy due to spastic muscles and the stress and strain on parts of the body that are compensating for muscle abnormalities. Some individuals may also have frequent and irregular muscle spasms that can’t be predicted or medicated in advance. Doctors often prescribe diazepam to reduce the pain associated with muscle spasms, but it’s not known exactly how the drug works to interfere with pain signals. The drug gabapentin has been used successfully to decrease the severity and frequency of painful spasms. BT-A injections have also been shown to decrease spasticity and pain, and are commonly given under anesthesia to avoid the pain associated with the injections. Intrathecal baclofen has shown good results in reducing pain, but its delivery is invasive, time intensive, and expensive. Some children and adults have been able to decrease pain by using noninvasive and drug-free interventions such as distraction, relaxation training, biofeedback, and therapeutic massage. Do Adults with Cerebral Palsy Face Special Health Challenges?Before the mid-twentieth century, few children with cerebral palsy survived to adulthood. Now, because of improvements in medical care, rehabilitation, and assistive technologies, 65 to 90 percent of children with cerebral palsy live into their adult years. This increase in life expectancy is often accompanied by a rise in medical and functional problems – some of them beginning at a relatively early age – including the following: Premature aging. The majority of individuals with cerebral palsy will experience some form of premature aging by the time they reach their 40s because of the extra stress and strain the disease puts upon their bodies. The developmental delays that often accompany cerebral palsy keep some organ systems from developing to their full capacity and level of performance. As a consequence, organ systems such as the cardiovascular system (the heart, veins, and arteries) and pulmonary system (lungs) have to work harder and they age prematurely. Functional issues at work. The day-to-day challenges of the workplace are likely to increase as an employed individual with cerebral palsy reaches middle age. Some individuals will be able to continue working with accommodations such as an adjusted work schedule, assistive equipment, or frequent rest periods. Early retirement may be necessary for others. Depression. Mental health issues can also be of concern as someone with cerebral palsy grows older. The rate of depression is three to four times higher in people with disabilities such as cerebral palsy. It appears to be related not so much to the severity of their disabilities, but to how well they cope with them. The amount of emotional support someone has, how successful they are at coping with disappointment and stress, and whether or not they have an optimistic outlook about the future all have a significant impact on mental health. Post-impairment syndrome. Most adults with cerebral palsy experience what is called post-impairment syndrome, a combination of pain, fatigue, and weakness due to muscle abnormalities, bone deformities, overuse syndromes (sometimes also called repetitive motion injuries), and arthritis. Fatigue is often a challenge, since individuals with cerebral palsy use three to five times the amount of energy that able-bodied people use when they walk and move about. Osteoarthritis and degenerative arthritis. Musculoskeletal abnormalities that may not produce discomfort during childhood can cause pain in adulthood. For example, the abnormal relationships between joint surfaces and excessive joint compression can lead to the early development of painful osteoarthritis and degenerative arthritis. Individuals with cerebral palsy also have limited strength and restricted patterns of movement, which puts them at risk for overuse syndromes and nerve entrapments. Pain. Issues related to pain often go unrecognized by health care providers since individuals with cerebral palsy may not be able to describe the extent or location of their pain. Pain can be acute or chronic, and is experienced most commonly in the hips, knees, ankles, and the upper and lower back. Individuals with spastic cerebral palsy have an increased number of painful sites and worse pain than those with other types of cerebral palsy. The best treatment for pain due to musculoskeletal abnormalities is preventive – correcting skeletal and muscle abnormalities early in life to avoid the progressive accumulation of stress and strain that causes pain. Dislocated hips, which are particularly likely to cause pain, can be surgically repaired. If it is managed properly, pain does not have to become a chronic condition. Other medical conditions. Adults have higher than normal rates of other medical conditions secondary to their cerebral palsy, such as hypertension, incontinence, bladder dysfunction, and swallowing difficulties. Curvature of the spine (scoliosis) is likely to progress after puberty, when bones have matured into their final shape and size. People with cerebral palsy also have a higher incidence of bone fractures, occurring most frequently during physical therapy sessions. A combination of mouth breathing, poor hygiene, and abnormalities in tooth enamel increase the risk of cavities and periodontal disease. Twenty-five percent to 39 percent of adults with cerebral palsy have vision problems; eight to 18 percent have hearing problems. Because of their unique medical situations, adults with cerebral palsy benefit from regular visits to their doctor and ongoing evaluation of their physical status. It is important to evaluate physical complaints to make sure they are not the result of underlying conditions. For example, adults with cerebral palsy are likely to experience fatigue, but fatigue can also be due to undiagnosed medical problems that could be treated and reversed. Because many individuals with cerebral palsy outlive their primary caregiver, the issue of long-term care and support should be taken into account and planned for. What Research Is Being Done?Investigators from many fields of medicine and health are using their expertise to help improve the treatment and diagnosis of cerebral palsy. Much of their work is supported through the NINDS, the National Institute of Child Health and Human Development (NICHD), other agencies within the federal government, nonprofit groups such as the United Cerebral Palsy Research and Educational Foundation, and other private institutions. The ultimate hope for curing cerebral palsy rests with prevention. In order to prevent cerebral palsy, however, scientists have to understand normal fetal brain development so that they can understand what happens when a baby’s brain develops abnormally. Between conception and the birth of a baby, one cell divides to form a handful of cells, and then hundreds, millions, and, eventually, billions of cells. Some of these cells specialize to become brain cells, and then specialize even further into particular types of neurons that travel to their appropriate place in the brain (a process that scientists call neuronal migration). Once they are in the right place, they establish connections with other brain cells. This is how the brain develops and becomes able to communicate with the rest of the body -- through overlapping neural circuits made up of billions of interconnected and interdependent neurons. Many scientists now think that a significant number of children develop cerebral palsy because of mishaps early in brain development. They are examining how brain cells specialize and form the right connections, and they are looking for ways to prevent the factors that disrupt the normal processes of brain development. Genetic defects are sometimes responsible for the brain malformations and abnormalities that cause cerebral palsy. Scientists funded by the NINDS are searching for the genes responsible for these abnormalities by collecting DNA samples from people with cerebral palsy and their families and using genetic screening techniques to discover linkages between individual genes and specific types of abnormality – primarily those associated with abnormal neuronal migration. Scientists are scrutinizing events in newborn babies’ brains, such as bleeding, epileptic seizures, and breathing and circulation problems, which can cause the abnormal release of chemicals that trigger the kind of damage that causes cerebral palsy. For example, research has shown that bleeding in the brain unleashes dangerously high amounts of a brain chemical called glutamate. Although glutamate is necessary in the brain to help neurons communicate, too much glutamate overexcites and kills neurons. Scientists are now looking closely at glutamate to detect how its release harms brain tissue. By learning how brain chemicals that are normally helpful become dangerously toxic, scientists will have opportunities to develop new drugs to block their harmful effects. Scientists funded by the NINDS are also investigating whether substances in the brain that protect neurons from damage, called neurotrophins, could be used to prevent brain damage as a result of stroke or oxygen deprivation. Understanding how these neuroprotective substances act would allow scientists to develop synthetic neurotrophins that could be given immediately after injury to prevent neuron death and damage. The relationship between uterine infections during pregnancy and the risk of cerebral palsy continues to be studied by researchers funded by the NIH. There is evidence that uterine infections trigger inflammation and the production of immune system cells called cytokines, which can pass into an unborn baby’s brain and interrupt normal development. By understanding what cytokines do in the fetal brain and the type of damage these immune system cells cause, researchers have the potential to develop medications that could be given to mothers with uterine infections to prevent brain damage in their unborn children. Approximately 10 percent of newborns are born prematurely, and of those babies, more than 10 percent will have brain injuries that will lead to cerebral palsy and other brain-based disabilities. A particular type of damage to the white matter of the brain, called periventricular leukomalacia (PVL), is the predominant form of brain injury in premature infants. NINDS-sponsored researchers studying PVL are looking for new strategies to prevent this kind of damage by developing safe, nontoxic therapies delivered to at-risk mothers to protect their unborn babies. Although congenital cerebral palsy is a condition that is present at birth, a year or two can pass before any disabilities are noticed. Researchers have shown that the earlier rehabilitative treatment begins, the better the outcome for children with cerebral palsy. But an early diagnosis is hampered by the lack of diagnostic techniques to identify brain damage or abnormalities in infants. Research funded by the NINDS is using imaging techniques, devices that measure electrical activity in the brain, and neurobehavioral tests to predict those preterm infants who will develop cerebral palsy. If these screening techniques are successful, doctors will be able to identify infants at risk for cerebral palsy before they are born. Noninvasive methods to record the brain activity of unborn babies in the womb and to identify those with brain damage or abnormalities would also be a valuable addition to the diagnostic tool kit. Another NINDS-funded study focuses on the development of fetal magnetoencephalography (fMEG) – a technology that would allow doctors to look for abnormalities in fetal brain activity. Epidemiological
studies – studies that look at the distribution and
causes of disease among people -- help scientists understand risk
factors and outcomes for particular diseases and medical
conditions.
Researchers have established that preterm birth (when a baby
is born before 32 weeks’ gestation) is the highest risk
factor for cerebral palsy. Consequently, the increasing
rate of premature births in the
While this research offers hope for preventing cerebral palsy in the future, ongoing research to improve treatment brightens the outlook for those who must face the challenges of cerebral palsy today. An important thrust of such research is the evaluation of treatments already in use so that physicians and parents have valid information to help them choose the best therapy. A good example of this effort is an ongoing NINDS-supported study that promises to yield new information about which patients are most likely to benefit from selective dorsal rhizotomy, a surgical technique that is increasingly being used to reduce spasticity (see Surgery). Similarly, although physical therapy programs are used almost universally to rehabilitate children with cerebral palsy, there are no definitive studies to indicate which techniques work best. For example, constraint-induced therapy (CIT) is a type of physical therapy that has been used successfully with adult stroke survivors and individuals who have traumatic brain injury and are left with a weak or disabled arm on one side of the body. The therapy involves restraining the stronger arm in a cast and forcing the weaker arm to perform 6 hours of intensive “shaping” activities every day over the course of 3 weeks. The researchers who conducted the clinical trials in adult stroke survivors realized CIT’s potential for strengthening children’s arms weakened by cerebral palsy. In a randomized, controlled study of children with cerebral palsy funded by the NIH, researchers put one group of children through conventional physical therapy and another group through 21 consecutive days of CIT. Researchers looked for evidence of improvement in the movement and function of the disabled arm, whether the improvement lasted after the end of treatment, and if it was associated with significant gains in other areas, such as trunk control, mobility, communication, and self-help skills. Children receiving CIT outperformed the children receiving conventional physical therapy across all measures of success, including how well they could move their arms after therapy and their ability to do new tasks during the study and then at home with their families. Six months later they still had better control of their arm. The results from this study are the first to prove the benefits of a physical therapy. Additional research to determine the optimal length and intensity of CIT will allow doctors to add this therapy to the cerebral palsy treatment toolbox. Studies have shown that functional electrical stimulation is an effective way to target and strengthen spastic muscles, but the method of delivering the electrical pulses requires expensive, bulky devices implanted by a surgeon, or skin surface stimulation applied by a trained therapist. NINDS-funded researchers have developed a high-tech method that does away with the bulky apparatus and lead wires by using a hypodermic needle to inject microscopic wireless devices into specific muscles or nerves. The devices are powered by a telemetry wand that can direct the number and strength of their pulses by remote control. The device has been used to activate and strengthen muscles in the hand, shoulder, and ankle in people with cerebral palsy as well as in stroke survivors. As researchers continue to explore new treatments for cerebral palsy and to expand our knowledge of brain development, we can expect significant improvements in the care of children with cerebral palsy and many other disorders that strike in early life. Where can I get more information? For more information on neurological disorders or research programs
funded by the National Institute of Neurological Disorders and
Stroke, contact the Institute's Brain Resources and Information
Network (BRAIN) at: BRAIN Information also is available from the following organizations:
acquired cerebral palsy — cerebral palsy that occurs as a result of injury to the brain after birth or during early childhood. Apgar score — a numbered scoring system doctors use to assess a baby's physical state at the time of birth. anticholinergic drugs — a family of drugs that inhibit parasympathetic neural activity by blocking the neurotransmitter acetylcholine. asphyxia — a lack of oxygen due to trouble with breathing or poor oxygen supply in the air. ataxia (ataxic) — the loss of muscle control. athetoid — making slow, sinuous, involuntary, writhing movements, especially with the hands. bilirubin — a bile pigment produced by the liver of the human body as a byproduct of digestion. bisphosphonates — a family of drugs that strengthen bones and reduce the risk of bone fracture in elderly adults. botulinum toxin — a drug commonly used to relax spastic muscles; it blocks the release of acetylcholine, a neurotransmitter that energizes muscle tissue. cerebral — relating to the two hemispheres of the human brain. cerebral dysgenesis — defective brain development. chemodenervation — a treatment that relaxes spastic muscles by interrupting nerve impulse pathways via a drug, such as botulinum toxin, which prevents communication between neurons and muscle tissue. choreoathetoid — a condition characterized by aimless muscle movements and involuntary motions. computed tomography (CT) scan — an imaging technique that uses X-rays and a computer to create a picture of the brain's tissues and structures. congenital cerebral palsy — cerebral palsy that is present at birth from causes that have occurred during fetal development. contracture — a condition in which muscles become fixed in a rigid, abnormal position, which causes distortion or deformity. cytokines — messenger cells that play a role in the inflammatory response to infection. developmental delay — behind schedule in reaching the milestones of early childhood development. disuse atrophy — muscle wasting caused by the inability to flex and exercise muscles. dyskinetic — the impairment of the ability to perform voluntary movements, which results in awkward or incomplete movements. dystonia (dystonic) a condition of abnormal muscle tone. electroencephalogram (EEG) — a technique for recording the pattern of electrical currents inside the brain. electromyography — a special recording technique that detects muscle activity. failure to thrive — a condition characterized by a lag in physical growth and development. focal (partial) seizure — a brief and temporary alteration in movement, sensation, or autonomic nerve function caused by abnormal electrical activity in a localized area of the brain. gait analysis — a technique that uses cameras, force plates, electromyography, and computer analysis to objectively measure an individual's pattern of walking. gastroesophageal reflux disease (GERD) — also known as heartburn, which happens when stomach acids back up into the esophagus. gastrostomy — a surgical procedure that creates an artificial opening in the stomach for the insertion of a feeding tube. gestation — the period of fetal development from the time of conception until birth. hemianopia — defective vision or blindness that impairs half of the normal field of vision. hemiparesis — paralysis affecting only one side of the body. homonymous — having the same description, name, or term. hypertonia — increased muscle tone. hypotonia — decreased muscle tone. hypoxic-ischemic encephalopathy — brain damage caused by poor blood flow or insufficient oxygen supply to the brain. intracranial hemorrhage — bleeding in the brain. intrapartum asphyxia — the reduction or total stoppage of oxygen circulating in a baby’s brain during labor and delivery. intrathecal
baclofen — baclofen that is injected into the
cerebrospinal fluid of the spinal cord to reduce spasticity. jaundice — a blood disorder caused by the abnormal buildup of bilirubin in the bloodstream. kernicterus — a neurological syndrome caused by deposition of bilirubin into brain tissues. Kernicterus develops in extremely jaundiced infants, especially those with severe Rh incompatibility. kyphosis — a humpback-like outward curvature of the upper spine. lordosis — an increased inward curvature of the lower spine. magnetic resonance imaging (MRI) — an imaging technique that uses radio waves, magnetic fields, and computer analysis to create a picture of body tissues and structures. nerve entrapment — repeated or prolonged pressure on a nerve root or peripheral nerve. neuronal migration — the process in the developing brain in which neurons migrate from where they are born to where they settle into neural circuits. Neuronal migration, which occurs as early as the second month of gestation, is controlled in the brain by chemical guides and signals. neuroprotective — describes substances that protect nervous system cells from damage or death. neurotrophins — a family of molecules that encourage survival of nervous system cells. off-label drugs — drugs prescribed to treat conditions other than those that have been approved by the Food and Drug Administration. orthotic devices — special devices, such as splints or braces, used to treat posture problems involving the muscles, ligaments, or bones. osteopenia — reduced density and mass of the bones. overuse syndrome (also called repetitive strain injury) — a condition in which repetitive movements or constrained posture cause nerve and muscle damage, which results in discomfort or persistent pain in muscles, tendons, and other soft tissues. This can happen in various parts of the body, but is most likely to happen in the arms, legs, or hands. palsy — paralysis, or the lack of control over voluntary movement. -paresis or -plegia — weakness or paralysis. In cerebral palsy, these terms are typically combined with other phrases that describe the distribution of paralysis and weakness; for example, quadriplegia means paralysis of all four limbs. pelvic inflammatory disease (PID, also sometimes called pelvic infection or intrauterine infection) — an infection of the upper genital tract (the uterus, ovaries, and fallopian tubes) caused by sexually transmitted infectious microorganisms. Symptoms of PID include fever, foul-smelling vaginal discharge, abdominal pain and pain during intercourse, and vaginal bleeding. Many different organisms can cause PID, but most cases are associated with gonorrhea and chlamydia. periventricular leukomalacia (PVL) — “peri" means near; "ventricular" refers to the ventricles or fluid spaces of the brain; and "leukomalacia" refers to softening of the white matter of the brain. PVL is a condition in which the cells that make up white matter die near the ventricles. Under a microscope, the tissue looks soft and sponge-like. placenta — an organ that joins a mother with her unborn baby and provides nourishment and sustenance. post-impairment syndrome — a combination of pain, fatigue, and weakness due to muscle abnormalities, bone deformities, overuse syndromes, or arthritis. quadriplegia — paralysis of both the arms and legs. respite care — rest or relief from caretaking obligations. Rh incompatibility — a blood condition in which antibodies in a pregnant woman's blood attack fetal blood cells and impair an unborn baby’s supply of oxygen and nutrients. rubella — (also known as German measles) a viral infection that can damage the nervous system of an unborn baby if a mother contracts the disease during pregnancy. scoliosis — a disease of the spine in which the spinal column tilts or curves to one side of the body. selective dorsal rhizotomy — a surgical procedure in which selected nerves are severed to reduce spasticity in the legs. selective vulnerability — a term that describes why some neurons are more vulnerable than others to particular diseases or conditions. For example, motor neurons are selectively vulnerable to the loss or reduction in levels of the neurotransmitter dopamine, which results in the weakness and paralysis of amyotrophic lateral sclerosis (ALS, commonly called Lou Gehrig’s disease). spastic (or spasticity) — describes stiff muscles and awkward movements. spastic diplegia (or diparesis) — a form of cerebral palsy in which spasticity affects both legs, but the arms are relatively or completely spared. spastic hemiplegia (or hemiparesis) — a form of cerebral palsy in which spasticity affects an arm and leg on one side of the body. spastic quadriplegia (or quadriparesis) — a form of cerebral palsy in which all four limbs are paralyzed or weakened equally. stereognosia — difficulty perceiving and identifying objects using the sense of touch. strabismus — misalignment of the eyes, also known as cross eyes. telemetry wand — a hand-held device that acts as a remote control, directing the dosing level of a drug via a pump implanted beneath the skin. tonic-clonic seizure — a type of seizure that results in loss of consciousness, generalized convulsions, loss of bladder control, and tongue biting followed by confusion and lethargy when the convulsions end. tremor — an involuntary trembling or quivering. ultrasound — a technique that bounces sound waves off tissue and bone and uses the pattern of echoes to form an image, called a sonogram. NIH Publication No. 06-159
|
|
Introduction:
The Universal Disorder
A Brief History of Pain The Two Faces of Pain: Acute and Chronic The A to Z of Pain How is Pain Diagnosed? How is Pain Treated? What is the Role of Age and Gender in Pain? Gender and Pain Pain in Aging and Pediatric Populations: Special Needs and Concerns A Pain Primer: What Do We Know About Pain? What is the Future of Pain Research? Where can I get more information? Appendix Spine Basics: The Vertebrae, Discs, and Spinal Cord The Nervous Systems Phantom Pain: How Does the Brain Feel? Chili Peppers, Capsaicin, and Pain Marijuana Nerve Blocks Introduction: The Universal DisorderYou know it at once. It may be the fiery sensation of a burn moments after your finger touches the stove. Or it's a dull ache above your brow after a day of stress and tension. Or you may recognize it as a sharp pierce in your back after you lift something heavy. It is pain. In its most benign form, it warns us that something isn't quite right, that we should take medicine or see a doctor. At its worst, however, pain robs us of our productivity, our well-being, and, for many of us suffering from extended illness, our very lives. Pain is a complex perception that differs enormously among individual patients, even those who appear to have identical injuries or illnesses. In 1931, the French medical missionary Dr. Albert Schweitzer wrote, "Pain is a more terrible lord of mankind than even death itself." Today, pain has become the universal disorder, a serious and costly public health issue, and a challenge for family, friends, and health care providers who must give support to the individual suffering from the physical as well as the emotional consequences of pain. A Brief History of PainAncient civilizations recorded on stone tablets accounts of pain and the treatments used: pressure, heat, water, and sun. Early humans related pain to evil, magic, and demons. Relief of pain was the responsibility of sorcerers, shamans, priests, and priestesses, who used herbs, rites, and ceremonies as their treatments. The Greeks and Romans were the first to advance a theory of sensation, the idea that the brain and nervous system have a role in producing the perception of pain. But it was not until the Middle Ages and well into the Renaissance-the 1400s and 1500s-that evidence began to accumulate in support of these theories. Leonardo da Vinci and his contemporaries came to believe that the brain was the central organ responsible for sensation. Da Vinci also developed the idea that the spinal cord transmits sensations to the brain. In the 17th and 18th centuries, the study of the body-and the senses-continued to be a source of wonder for the world's philosophers. In 1664, the French philosopher René Descartes described what to this day is still called a "pain pathway." Descartes illustrated how particles of fire, in contact with the foot, travel to the brain and he compared pain sensation to the ringing of a bell. In the 19th century, pain came to dwell under a new domain-science-paving the way for advances in pain therapy. Physician-scientists discovered that opium, morphine, codeine, and cocaine could be used to treat pain. These drugs led to the development of aspirin, to this day the most commonly used pain reliever. Before long, anesthesia-both general and regional-was refined and applied during surgery. "It has no future but itself," wrote the 19th century American poet Emily Dickinson, speaking about pain. As the 21st century unfolds, however, advances in pain research are creating a less grim future than that portrayed in Dickinson’s verse, a future that includes a better understanding of pain, along with greatly improved treatments to keep it in check. The Two Faces of Pain: Acute and ChronicWhat is pain? The International Association for the Study of Pain defines it as: An unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage. It is useful to distinguish between two basic types of pain, acute and chronic, and they differ greatly.
The A to Z of PainHundreds of pain syndromes or disorders make up the spectrum of pain. There are the most benign, fleeting sensations of pain, such as a pin prick. There is the pain of childbirth, the pain of a heart attack, and the pain that sometimes follows amputation of a limb. There is also pain accompanying cancer and the pain that follows severe trauma, such as that associated with head and spinal cord injuries. A sampling of common pain syndromes follows, listed alphabetically. Arachnoiditis is a condition in which one of the three membranes covering the brain and spinal cord, called the arachnoid membrane, becomes inflamed. A number of causes, including infection or trauma, can result in inflammation of this membrane. Arachnoiditis can produce disabling, progressive, and even permanent pain. Arthritis. Millions of Americans suffer from arthritic conditions such as osteoarthritis, rheumatoid arthritis, ankylosing spondylitis, and gout. These disorders are characterized by joint pain in the extremities. Many other inflammatory diseases affect the body's soft tissues, including tendonitis and bursitis. Back pain has become the high price paid by our modern lifestyle and is a startlingly common cause of disability for many Americans, including both active and inactive people. Back pain that spreads to the leg is called sciatica and is a very common condition (see below). Another common type of back pain is associated with the discs of the spine, the soft, spongy padding between the vertebrae (bones) that form the spine. Discs protect the spine by absorbing shock, but they tend to degenerate over time and may sometimes rupture. Spondylolisthesis is a back condition that occurs when one vertebra extends over another, causing pressure on nerves and therefore pain. Also, damage to nerve roots (see Spine Basics in the Appendix) is a serious condition, called radiculopathy, that can be extremely painful. Treatment for a damaged disc includes drugs such as painkillers, muscle relaxants, and steroids; exercise or rest, depending on the patient's condition; adequate support, such as a brace or better mattress and physical therapy. In some cases, surgery may be required to remove the damaged portion of the disc and return it to its previous condition, especially when it is pressing a nerve root. Surgical procedures include discectomy, laminectomy, or spinal fusion (see section on surgery in How is Pain Treated? for more information on these treatments). Burn pain can be profound and poses an extreme challenge to the medical community. First-degree burns are the least severe; with third-degree burns, the skin is lost. Depending on the injury, pain accompanying burns can be excruciating, and even after the wound has healed patients may have chronic pain at the burn site. Central pain syndrome-see "Trauma" below. Cancer pain can accompany the growth of a tumor, the treatment of cancer, or chronic problems related to cancer's permanent effects on the body. Fortunately, most cancer pain can be treated to help minimize discomfort and stress to the patient. Headaches affect millions of Americans. The three most common types of chronic headache are migraines, cluster headaches, and tension headaches. Each comes with its own telltale brand of pain.
Head and facial pain can be agonizing, whether it results from dental problems or from disorders such as cranial neuralgia, in which one of the nerves in the face, head, or neck is inflamed. Another condition, trigeminal neuralgia (also called tic douloureux), affects the largest of the cranial nerves (see The Nervous Systems in the Appendix) and is characterized by a stabbing, shooting pain. Muscle pain can range from an aching muscle, spasm, or strain, to the severe spasticity that accompanies paralysis. Another disabling syndrome is fibromyalgia, a disorder characterized by fatigue, stiffness, joint tenderness, and widespread muscle pain. Polymyositis, dermatomyositis, and inclusion body myositis are painful disorders characterized by muscle inflammation. They may be caused by infection or autoimmune dysfunction and are sometimes associated with connective tissue disorders, such as lupus and rheumatoid arthritis. Myofascial pain syndromes affect sensitive areas known as trigger points, located within the body's muscles. Myofascial pain syndromes are sometimes misdiagnosed and can be debilitating. Fibromyalgia is a type of myofascial pain syndrome. Neuropathic pain is a type of pain that can result from injury to nerves, either in the peripheral or central nervous system (see The Nervous Systems in the Appendix). Neuropathic pain can occur in any part of the body and is frequently described as a hot, burning sensation, which can be devastating to the affected individual. It can result from diseases that affect nerves (such as diabetes) or from trauma, or, because chemotherapy drugs can affect nerves, it can be a consequence of cancer treatment. Among the many neuropathic pain conditions are diabetic neuropathy (which results from nerve damage secondary to vascular problems that occur with diabetes); reflex sympathetic dystrophy syndrome (see below), which can follow injury; phantom limb and post-amputation pain (see Phantom Pain in the Appendix), which can result from the surgical removal of a limb; postherpetic neuralgia, which can occur after an outbreak of shingles; and central pain syndrome, which can result from trauma to the brain or spinal cord. Reflex sympathetic dystrophy syndrome, or RSDS, is accompanied by burning pain and hypersensitivity to temperature. Often triggered by trauma or nerve damage, RSDS causes the skin of the affected area to become characteristically shiny. In recent years, RSDS has come to be called complex regional pain syndrome (CRPS); in the past it was often called causalgia. Repetitive stress injuries are muscular conditions that result from repeated motions performed in the course of normal work or other daily activities. They include:
Sciatica is a painful condition caused by pressure on the sciatic nerve, the main nerve that branches off the spinal cord and continues down into the thighs, legs, ankles, and feet. Sciatica is characterized by pain in the buttocks and can be caused by a number of factors. Exertion, obesity, and poor posture can all cause pressure on the sciatic nerve. One common cause of sciatica is a herniated disc (see Spine Basics in the Appendix). Shingles and other painful disorders affect the skin. Pain is a common symptom of many skin disorders, even the most common rashes. One of the most vexing neurological disorders is shingles or herpes zoster, an infection that often causes agonizing pain resistant to treatment. Prompt treatment with antiviral agents is important to arrest the infection, which if prolonged can result in an associated condition known as postherpetic neuralgia. Other painful disorders affecting the skin include:
Sports injuries are common. Sprains, strains, bruises, dislocations, and fractures are all well-known words in the language of sports. Pain is another. In extreme cases, sports injuries can take the form of costly and painful spinal cord and head injuries, which cause severe suffering and disability. Spinal stenosis refers to a narrowing of the canal surrounding the spinal cord. The condition occurs naturally with aging. Spinal stenosis causes weakness in the legs and leg pain usually felt while the person is standing up and often relieved by sitting down. Surgical pain may require regional or general anesthesia during the procedure and medications to control discomfort following the operation. Control of pain associated with surgery includes presurgical preparation and careful monitoring of the patient during and after the procedure. Temporomandibular disorders are conditions in which the temporomandibular joint (the jaw) is damaged and/or the muscles used for chewing and talking become stressed, causing pain. The condition may be the result of a number of factors, such as an injury to the jaw or joint misalignment, and may give rise to a variety of symptoms, most commonly pain in the jaw, face, and/or neck muscles. Physicians reach a diagnosis by listening to the patient's description of the symptoms and by performing a simple examination of the facial muscles and the temporomandibular joint. Trauma can occur after injuries in the home, at the workplace, during sports activities, or on the road. Any of these injuries can result in severe disability and pain. Some patients who have had an injury to the spinal cord experience intense pain ranging from tingling to burning and, commonly, both. Such patients are sensitive to hot and cold temperatures and touch. For these individuals, a touch can be perceived as intense burning, indicating abnormal signals relayed to and from the brain. This condition is called central pain syndrome or, if the damage is in the thalamus (the brain's center for processing bodily sensations), thalamic pain syndrome. It affects as many as 100,000 Americans with multiple sclerosis, Parkinson's disease, amputated limbs, spinal cord injuries, and stroke. Their pain is severe and is extremely difficult to treat effectively. A variety of medications, including analgesics, antidepressants, anticonvulsants, and electrical stimulation, are options available to central pain patients. Vascular disease or injury-such as vasculitis or inflammation of blood vessels, coronary artery disease, and circulatory problems-all have the potential to cause pain. Vascular pain affects millions of Americans and occurs when communication between blood vessels and nerves is interrupted. Ruptures, spasms, constriction, or obstruction of blood vessels, as well as a condition called ischemia in which blood supply to organs, tissues, or limbs is cut off, can also result in pain. How is Pain Diagnosed?There is no way to tell how much pain a person has. No test can measure the intensity of pain, no imaging device can show pain, and no instrument can locate pain precisely. Sometimes, as in the case of headaches, physicians find that the best aid to diagnosis is the patient's own description of the type, duration, and location of pain. Defining pain as sharp or dull, constant or intermittent, burning or aching may give the best clues to the cause of pain. These descriptions are part of what is called the pain history, taken by the physician during the preliminary examination of a patient with pain. Physicians, however, do have a number of technologies they use to find the cause of pain. Primarily these include:
How is Pain Treated?The goal of pain management is to improve function, enabling individuals to work, attend school, or participate in other day-to-day activities. Patients and their physicians have a number of options for the treatment of pain; some are more effective than others. Sometimes, relaxation and the use of imagery as a distraction provide relief. These methods can be powerful and effective, according to those who advocate their use. Whatever the treatment regime, it is important to remember that pain is treatable. The following treatments are among the most common. Acetaminophen is the basic ingredient found in Tylenol® and its many generic equivalents. It is sold over the counter, in a prescription-strength preparation, and in combination with codeine (also by prescription). Acupuncture dates back 2,500 years and involves the application of needles to precise points on the body. It is part of a general category of healing called traditional Chinese or Oriental medicine. Acupuncture remains controversial but is quite popular and may one day prove to be useful for a variety of conditions as it continues to be explored by practitioners, patients, and investigators. Analgesic refers to the class of drugs that includes most painkillers, such as aspirin, acetaminophen, and ibuprofen. The word analgesic is derived from ancient Greek and means to reduce or stop pain. Nonprescription or over-the-counter pain relievers are generally used for mild to moderate pain. Prescription pain relievers, sold through a pharmacy under the direction of a physician, are used for more moderate to severe pain. Anticonvulsants are used for the treatment of seizure disorders but are also sometimes prescribed for the treatment of pain. Carbamazepine in particular is used to treat a number of painful conditions, including trigeminal neuralgia. Another antiepileptic drug, gabapentin, is being studied for its pain-relieving properties, especially as a treatment for neuropathic pain. Antidepressants are sometimes used for the treatment of pain and, along with neuroleptics and lithium, belong to a category of drugs called psychotropic drugs. In addition, anti-anxiety drugs called benzodiazepines also act as muscle relaxants and are sometimes used as pain relievers. Physicians usually try to treat the condition with analgesics before prescribing these drugs. Antimigraine drugs include the triptans- sumatriptan (Imitrex®), naratriptan (Amerge®), and zolmitriptan (Zomig®)-and are used specifically for migraine headaches. They can have serious side effects in some people and therefore, as with all prescription medicines, should be used only under a doctor's care. Aspirin may be the most widely used pain-relief agent and has been sold over the counter since 1905 as a treatment for fever, headache, and muscle soreness. Biofeedback is used for the treatment of many common pain problems, most notably headache and back pain. Using a special electronic machine, the patient is trained to become aware of, to follow, and to gain control over certain bodily functions, including muscle tension, heart rate, and skin temperature. The individual can then learn to effect a change in his or her responses to pain, for example, by using relaxation techniques. Biofeedback is often used in combination with other treatment methods, generally without side effects. Similarly, the use of relaxation techniques in the treatment of pain can increase the patient's feeling of well-being. Capsaicin is a chemical found in chili peppers that is also a primary ingredient in pain-relieving creams (see Chili Peppers, Capsaicin, and Pain in the Appendix). Chemonucleolysis is a treatment in which an enzyme, chymopapain, is injected directly into a herniated lumbar disc (see Spine Basics in the Appendix) in an effort to dissolve material around the disc, thus reducing pressure and pain. The procedure's use is extremely limited, in part because some patients may have a life-threatening allergic reaction to chymopapain. Chiropractic care may ease back pain, neck pain, headaches, and musculoskeletal conditions. It involves "hands-on" therapy designed to adjust the relationship between the body's structure (mainly the spine) and its functioning. Chiropractic spinal manipulation includes the adjustment and manipulation of the joints and adjacent tissues. Such care may also involve therapeutic and rehabilitative exercises. Cognitive-behavioral therapy involves a wide variety of coping skills and relaxation methods to help prepare for and cope with pain. It is used for postoperative pain, cancer pain, and the pain of childbirth. Counseling can give a patient suffering from pain much needed support, whether it is derived from family, group, or individual counseling. Support groups can provide an important adjunct to drug or surgical treatment. Psychological treatment can also help patients learn about the physiological changes produced by pain. COX-2 inhibitors may be effective for individuals with arthritis. For many years scientists have wanted to develop a drug that works as well as morphine but without its negative side effects. Nonsteroidal anti-inflammatory drugs (NSAIDs) work by blocking two enzymes, cyclooxygenase-1 and cyclooxygenase-2, both of which promote production of hormones called prostaglandins, which in turn cause inflammation, fever, and pain. The newer COX-2 inhibitors primarily block cyclooxygenase-2 and are less likely to have the gastrointestinal side effects sometimes produced by NSAIDs. In 1999, the Food and Drug Administration approved a COX-2 inhibitor-celecoxib-for use in cases of chronic pain. The long-term effects of all COX-2 inhibitors are still being evaluated, especially in light of new information suggesting that these drugs may increase the risk of heart attack and stroke. Patients taking any of the COX-2 inhibitors should review their drug treatment with their doctors. Electrical stimulation, including transcutaneous electrical stimulation (TENS), implanted electric nerve stimulation, and deep brain or spinal cord stimulation, is the modern-day extension of age-old practices in which the nerves of muscles are subjected to a variety of stimuli, including heat or massage. Electrical stimulation, no matter what form, involves a major surgical procedure and is not for everyone, nor is it 100 percent effective. The following techniques each require specialized equipment and personnel trained in the specific procedure being used:
Exercise has come to be a prescribed part of some doctors' treatment regimes for patients with pain. Because there is a known link between many types of chronic pain and tense, weak muscles, exercise-even light to moderate exercise such as walking or swimming-can contribute to an overall sense of well-being by improving blood and oxygen flow to muscles. Just as we know that stress contributes to pain, we also know that exercise, sleep, and relaxation can all help reduce stress, thereby helping to alleviate pain. Exercise has been proven to help many people with low back pain. It is important, however, that patients carefully follow the routine laid out by their physicians. Hypnosis, first approved for medical use by the American Medical Association in 1958, continues to grow in popularity, especially as an adjunct to pain medication. In general, hypnosis is used to control physical function or response, that is, the amount of pain an individual can withstand. How hypnosis works is not fully understood. Some believe that hypnosis delivers the patient into a trance-like state, while others feel that the individual is simply better able to concentrate and relax or is more responsive to suggestion. Hypnosis may result in relief of pain by acting on chemicals in the nervous system, slowing impulses. Whether and how hypnosis works involves greater insight-and research-into the mechanisms underlying human consciousness. Ibuprofen is a member of the aspirin family of analgesics, the so-called nonsteroidal anti-inflammatory drugs (see below). It is sold over the counter and also comes in prescription-strength preparations. Low-power lasers have been used occasionally by some physical therapists as a treatment for pain, but like many other treatments, this method is not without controversy. Magnets are increasingly popular with athletes who swear by their effectiveness for the control of sports-related pain and other painful conditions. Usually worn as a collar or wristwatch, the use of magnets as a treatment dates back to the ancient Egyptians and Greeks. While it is often dismissed as quackery and pseudoscience by skeptics, proponents offer the theory that magnets may effect changes in cells or body chemistry, thus producing pain relief. Narcotics (see Opioids, below). Nerve blocks employ the use of drugs, chemical agents, or surgical techniques to interrupt the relay of pain messages between specific areas of the body and the brain. There are many different names for the procedure, depending on the technique or agent used. Types of surgical nerve blocks include neurectomy; spinal dorsal, cranial, and trigeminal rhizotomy; and sympathectomy, also called sympathetic blockade (see Nerve Blocks in the Appendix). Nonsteroidal anti-inflammatory drugs (NSAIDs) (including aspirin and ibuprofen) are widely prescribed and sometimes called non-narcotic or non-opioid analgesics. They work by reducing inflammatory responses in tissues. Many of these drugs irritate the stomach and for that reason are usually taken with food. Although acetaminophen may have some anti-inflammatory effects, it is generally distinguished from the traditional NSAIDs. Opioids are derived from the poppy plant and are among the oldest drugs known to humankind. They include codeine and perhaps the most well-known narcotic of all, morphine. Morphine can be administered in a variety of forms, including a pump for patient self-administration. Opioids have a narcotic effect, that is, they induce sedation as well as pain relief, and some patients may become physically dependent upon them. For these reasons, patients given opioids should be monitored carefully; in some cases stimulants may be prescribed to counteract the sedative side effects. In addition to drowsiness, other common side effects include constipation, nausea, and vomiting. Physical therapy and rehabilitation date back to the ancient practice of using physical techniques and methods, such as heat, cold, exercise, massage, and manipulation, in the treatment of certain conditions. These may be applied to increase function, control pain, and speed the patient toward full recovery. Placebos offer some individuals pain relief although whether and how they have an effect is mysterious and somewhat controversial. Placebos are inactive substances, such as sugar pills, or harmless procedures, such as saline injections or sham surgeries, generally used in clinical studies as control factors to help determine the efficacy of active treatments. Although placebos have no direct effect on the underlying causes of pain, evidence from clinical studies suggests that many pain conditions such as migraine headache, back pain, post-surgical pain, rheumatoid arthritis, angina, and depression sometimes respond well to them. This positive response is known as the placebo effect, which is defined as the observable or measurable change that can occur in patients after administration of a placebo. Some experts believe the effect is psychological and that placebos work because the patients believe or expect them to work. Others say placebos relieve pain by stimulating the brain's own analgesics and setting the body's self-healing forces in motion. A third theory suggests that the act of taking placebos relieves stress and anxiety-which are known to aggravate some painful conditions-and, thus, cause the patients to feel better. Still, placebos are considered controversial because by definition they are inactive and have no actual curative value. R.I.C.E.-Rest, Ice, Compression, and Elevation-are four components prescribed by many orthopedists, coaches, trainers, nurses, and other professionals for temporary muscle or joint conditions, such as sprains or strains. While many common orthopedic problems can be controlled with these four simple steps, especially when combined with over-the-counter pain relievers, more serious conditions may require surgery or physical therapy, including exercise, joint movement or manipulation, and stimulation of muscles. Surgery, although not always an option, may be required to relieve pain, especially pain caused by back problems or serious musculoskeletal injuries. Surgery may take the form of a nerve block (see Nerve Blocks in the Appendix) or it may involve an operation to relieve pain from a ruptured disc. Surgical procedures for back problems include discectomy or, when microsurgical techniques are used, microdiscectomy, in which the entire disc is removed; laminectomy, a procedure in which a surgeon removes only a disc fragment, gaining access by entering through the arched portion of a vertebra; and spinal fusion, a procedure where the entire disc is removed and replaced with a bone graft. In a spinal fusion, the two vertebrae are then fused together. Although the operation can cause the spine to stiffen, resulting in lost flexibility, the procedure serves one critical purpose: protection of the spinal cord. Other operations for pain include rhizotomy, in which a nerve close to the spinal cord is cut, and cordotomy, where bundles of nerves within the spinal cord are severed. Cordotomy is generally used only for the pain of terminal cancer that does not respond to other therapies. Another operation for pain is the dorsal root entry zone operation, or DREZ, in which spinal neurons corresponding to the patient's pain are destroyed surgically. Because surgery can result in scar tissue formation that may cause additional problems, patients are well advised to seek a second opinion before proceeding. Occasionally, surgery is carried out with electrodes that selectively damage neurons in a targeted area of the brain. These procedures rarely result in long-term pain relief, but both physician and patient may decide that the surgical procedure will be effective enough that it justifies the expense and risk. In some cases, the results of an operation are remarkable. For example, many individuals suffering from trigeminal neuralgia who are not responsive to drug treatment have had great success with a procedure called microvascular decompression, in which tiny blood vessels are surgically separated from surrounding nerves. What is the Role of Age and Gender in Pain?Gender and PainIt is now widely believed that pain affects men and women differently. While the sex hormones estrogen and testosterone certainly play a role in this phenomenon, psychology and culture, too, may account at least in part for differences in how men and women receive pain signals. For example, young children may learn to respond to pain based on how they are treated when they experience pain. Some children may be cuddled and comforted, while others may be encouraged to tough it out and to dismiss their pain. Many investigators are turning their attention to the study of gender differences and pain. Women, many experts now agree, recover more quickly from pain, seek help more quickly for their pain, and are less likely to allow pain to control their lives. They also are more likely to marshal a variety of resources-coping skills, support, and distraction-with which to deal with their pain. Research in this area is yielding fascinating results. For example, male experimental animals injected with estrogen, a female sex hormone, appear to have a lower tolerance for pain-that is, the addition of estrogen appears to lower the pain threshold. Similarly, the presence of testosterone, a male hormone, appears to elevate tolerance for pain in female mice: the animals are simply able to withstand pain better. Female mice deprived of estrogen during experiments react to stress similarly to male animals. Estrogen, therefore, may act as a sort of pain switch, turning on the ability to recognize pain. Investigators know that males and females both have strong natural pain-killing systems, but these systems operate differently. For example, a class of painkillers called kappa-opioids is named after one of several opioid receptors to which they bind, the kappa-opioid receptor, and they include the compounds nalbuphine (Nubain®) and butorphanol (Stadol®). Research suggests that kappa-opioids provide better pain relief in women. Though not prescribed widely, kappa-opioids are currently used for relief of labor pain and in general work best for short-term pain. Investigators are not certain why kappa-opioids work better in women than men. Is it because a woman's estrogen makes them work, or because a man's testosterone prevents them from working? Or is there another explanation, such as differences between men and women in their perception of pain? Continued research may result in a better understanding of how pain affects women differently from men, enabling new and better pain medications to be designed with gender in mind. Pain in Aging and Pediatric Populations: Special Needs and ConcernsPain is the number one complaint of older Americans, and one in five older Americans takes a painkiller regularly. In 1998, the American Geriatrics Society (AGS) issued guidelines* for the management of pain in older people. The AGS panel addressed the incorporation of several non-drug approaches in patients' treatment plans, including exercise. AGS panel members recommend that, whenever possible, patients use alternatives to aspirin, ibuprofen, and other NSAIDs because of the drugs' side effects, including stomach irritation and gastrointestinal bleeding. For older adults, acetaminophen is the first-line treatment for mild-to-moderate pain, according to the guidelines. More serious chronic pain conditions may require opioid drugs (narcotics), including codeine or morphine, for relief of pain. Pain in younger patients also requires special attention, particularly because young children are not always able to describe the degree of pain they are experiencing. Although treating pain in pediatric patients poses a special challenge to physicians and parents alike, pediatric patients should never be undertreated. Recently, special tools for measuring pain in children have been developed that, when combined with cues used by parents, help physicians select the most effective treatments. Nonsteroidal agents, and especially acetaminophen, are most often prescribed for control of pain in children. In the case of severe pain or pain following surgery, acetaminophen may be combined with codeine. * Journal of the American Geriatrics Society (1998; 46:635-651). A Pain Primer: What Do We Know About Pain?We may experience pain as a prick, tingle, sting, burn, or ache. Receptors on the skin trigger a series of events, beginning with an electrical impulse that travels from the skin to the spinal cord. The spinal cord acts as a sort of relay center where the pain signal can be blocked, enhanced, or otherwise modified before it is relayed to the brain. One area of the spinal cord in particular, called the dorsal horn (see section on Spine Basics in the Appendix), is important in the reception of pain signals. The most common destination in the brain for pain signals is the thalamus and from there to the cortex, the headquarters for complex thoughts. The thalamus also serves as the brain's storage area for images of the body and plays a key role in relaying messages between the brain and various parts of the body. In people who undergo an amputation, the representation of the amputated limb is stored in the thalamus. (For a discussion of the thalamus and its role in this phenomenon, called phantom pain, see section on Phantom Pain in the Appendix.) Pain is a complicated process that involves an intricate interplay between a number of important chemicals found naturally in the brain and spinal cord. In general, these chemicals, called neurotransmitters, transmit nerve impulses from one cell to another. There are many different neurotransmitters in the human body; some play a role in human disease and, in the case of pain, act in various combinations to produce painful sensations in the body. Some chemicals govern mild pain sensations; others control intense or severe pain. The body's chemicals act in the transmission of pain messages by stimulating neurotransmitter receptors found on the surface of cells; each receptor has a corresponding neurotransmitter. Receptors function much like gates or ports and enable pain messages to pass through and on to neighboring cells. One brain chemical of special interest to neuroscientists is glutamate. During experiments, mice with blocked glutamate receptors show a reduction in their responses to pain. Other important receptors in pain transmission are opiate-like receptors. Morphine and other opioid drugs work by locking on to these opioid receptors, switching on pain-inhibiting pathways or circuits, and thereby blocking pain. Another type of receptor that responds to painful stimuli is called a nociceptor. Nociceptors are thin nerve fibers in the skin, muscle, and other body tissues, that, when stimulated, carry pain signals to the spinal cord and brain. Normally, nociceptors only respond to strong stimuli such as a pinch. However, when tissues become injured or inflamed, as with a sunburn or infection, they release chemicals that make nociceptors much more sensitive and cause them to transmit pain signals in response to even gentle stimuli such as breeze or a caress. This condition is called allodynia -a state in which pain is produced by innocuous stimuli. The body's natural painkillers may yet prove to be the most promising pain relievers, pointing to one of the most important new avenues in drug development. The brain may signal the release of painkillers found in the spinal cord, including serotonin, norepinephrine, and opioid-like chemicals. Many pharmaceutical companies are working to synthesize these substances in laboratories as future medications. Endorphins and enkephalins are other natural painkillers. Endorphins may be responsible for the "feel good" effects experienced by many people after rigorous exercise; they are also implicated in the pleasurable effects of smoking. Similarly, peptides, compounds that make up proteins in the body, play a role in pain responses. Mice bred experimentally to lack a gene for two peptides called tachykinins-neurokinin A and substance P-have a reduced response to severe pain. When exposed to mild pain, these mice react in the same way as mice that carry the missing gene. But when exposed to more severe pain, the mice exhibit a reduced pain response. This suggests that the two peptides are involved in the production of pain sensations, especially moderate-to-severe pain. Continued research on tachykinins, conducted with support from the NINDS, may pave the way for drugs tailored to treat different severities of pain. Scientists are working to develop potent pain-killing drugs that act on receptors for the chemical acetylcholine. For example, a type of frog native to Ecuador has been found to have a chemical in its skin called epibatidine, derived from the frog's scientific name, Epipedobates tricolor. Although highly toxic, epibatidine is a potent analgesic and, surprisingly, resembles the chemical nicotine found in cigarettes. Also under development are other less toxic compounds that act on acetylcholine receptors and may prove to be more potent than morphine but without its addictive properties. The idea of using receptors as gateways for pain drugs is a novel idea, supported by experiments involving substance P. Investigators have been able to isolate a tiny population of neurons, located in the spinal cord, that together form a major portion of the pathway responsible for carrying persistent pain signals to the brain. When animals were given injections of a lethal cocktail containing substance P linked to the chemical saporin, this group of cells, whose sole function is to communicate pain, were killed. Receptors for substance P served as a portal or point of entry for the compound. Within days of the injections, the targeted neurons, located in the outer layer of the spinal cord along its entire length, absorbed the compound and were neutralized. The animals' behavior was completely normal; they no longer exhibited signs of pain following injury or had an exaggerated pain response. Importantly, the animals still responded to acute, that is, normal, pain. This is a critical finding as it is important to retain the body's ability to detect potentially injurious stimuli. The protective, early warning signal that pain provides is essential for normal functioning. If this work can be translated clinically, humans might be able to benefit from similar compounds introduced, for example, through lumbar (spinal) puncture. Another promising area of research using the body's natural pain-killing abilities is the transplantation of chromaffin cells into the spinal cords of animals bred experimentally to develop arthritis. Chromaffin cells produce several of the body's pain-killing substances and are part of the adrenal medulla, which sits on top of the kidney. Within a week or so, rats receiving these transplants cease to exhibit telltale signs of pain. Scientists, working with support from the NINDS, believe the transplants help the animals recover from pain-related cellular damage. Extensive animal studies will be required to learn if this technique might be of value to humans with severe pain. One way to control pain outside of the brain, that is, peripherally, is by inhibiting hormones called prostaglandins. Prostaglandins stimulate nerves at the site of injury and cause inflammation and fever. Certain drugs, including NSAIDs, act against such hormones by blocking the enzyme that is required for their synthesis. Blood vessel walls stretch or dilate during a migraine attack and it is thought that serotonin plays a complicated role in this process. For example, before a migraine headache, serotonin levels fall. Drugs for migraine include the triptans: sumatriptan (Imitrix®), naratriptan (Amerge®), and zolmitriptan (Zomig®). They are called serotonin agonists because they mimic the action of endogenous (natural) serotonin and bind to specific subtypes of serotonin receptors. Ongoing pain research, much of it supported by the NINDS, continues to reveal at an unprecedented pace fascinating insights into how genetics, the immune system, and the skin contribute to pain responses. The explosion of knowledge about human genetics is helping scientists who work in the field of drug development. We know, for example, that the pain-killing properties of codeine rely heavily on a liver enzyme, CYP2D6, which helps convert codeine into morphine. A small number of people genetically lack the enzyme CYP2D6; when given codeine, these individuals do not get pain relief. CYP2D6 also helps break down certain other drugs. People who genetically lack CYP2D6 may not be able to cleanse their systems of these drugs and may be vulnerable to drug toxicity. CYP2D6 is currently under investigation for its role in pain. In his research, the late John C. Liebeskind, a renowned pain expert and a professor of psychology at UCLA, found that pain can kill by delaying healing and causing cancer to spread. In his pioneering research on the immune system and pain, Dr. Liebeskind studied the effects of stress-such as surgery-on the immune system and in particular on cells called natural killer or NK cells. These cells are thought to help protect the body against tumors. In one study conducted with rats, Dr. Liebeskind found that, following experimental surgery, NK cell activity was suppressed, causing the cancer to spread more rapidly. When the animals were treated with morphine, however, they were able to avoid this reaction to stress. The link between the nervous and immune systems is an important one. Cytokines, a type of protein found in the nervous system, are also part of the body's immune system, the body's shield for fighting off disease. Cytokines can trigger pain by promoting inflammation, even in the absence of injury or damage. Certain types of cytokines have been linked to nervous system injury. After trauma, cytokine levels rise in the brain and spinal cord and at the site in the peripheral nervous system where the injury occurred. Improvements in our understanding of the precise role of cytokines in producing pain, especially pain resulting from injury, may lead to new classes of drugs that can block the action of these substances. What is the Future of Pain Research?In the forefront of pain research are scientists supported by the National Institutes of Health (NIH), including the NINDS. Other institutes at NIH that support pain research include the National Institute of Dental and Craniofacial Research, the National Cancer Institute, the National Institute of Nursing Research, the National Institute on Drug Abuse, and the National Institute of Mental Health. Developing better pain treatments is the primary goal of all pain research being conducted by these institutes. Some pain medications dull the patient's perception of pain. Morphine is one such drug. It works through the body's natural pain-killing machinery, preventing pain messages from reaching the brain. Scientists are working toward the development of a morphine-like drug that will have the pain-deadening qualities of morphine but without the drug's negative side effects, such as sedation and the potential for addiction. Patients receiving morphine also face the problem of morphine tolerance, meaning that over time they require higher doses of the drug to achieve the same pain relief. Studies have identified factors that contribute to the development of tolerance; continued progress in this line of research should eventually allow patients to take lower doses of morphine. One objective of investigators working to develop the future generation of pain medications is to take full advantage of the body's pain "switching center" by formulating compounds that will prevent pain signals from being amplified or stop them altogether. Blocking or interrupting pain signals, especially when there is no injury or trauma to tissue, is an important goal in the development of pain medications. An increased understanding of the basic mechanisms of pain will have profound implications for the development of future medicines. The following areas of research are bringing us closer to an ideal pain drug. Systems and Imaging: The idea of mapping cognitive functions to precise areas of the brain dates back to phrenology, the now archaic practice of studying bumps on the head. Positron emission tomography (PET), functional magnetic resonance imaging (fMRI), and other imaging technologies offer a vivid picture of what is happening in the brain as it processes pain. Using imaging, investigators can now see that pain activates at least three or four key areas of the brain's cortex-the layer of tissue that covers the brain. Interestingly, when patients undergo hypnosis so that the unpleasantness of a painful stimulus is not experienced, activity in some, but not all, brain areas is reduced. This emphasizes that the experience of pain involves a strong emotional component as well as the sensory experience, namely the intensity of the stimulus. Channels: The frontier in the search for new drug targets is represented by channels. Channels are gate-like passages found along the membranes of cells that allow electrically charged chemical particles called ions to pass into the cells. Ion channels are important for transmitting signals through the nerve's membrane. The possibility now exists for developing new classes of drugs, including pain cocktails that would act at the site of channel activity. Trophic Factors: A class of "rescuer" or "restorer" drugs may emerge from our growing knowledge of trophic factors, natural chemical substances found in the human body that affect the survival and function of cells. Trophic factors also promote cell death, but little is known about how something beneficial can become harmful. Investigators have observed that an over-accumulation of certain trophic factors in the nerve cells of animals results in heightened pain sensitivity, and that some receptors found on cells respond to trophic factors and interact with each other. These receptors may provide targets for new pain therapies. Molecular Genetics: Certain genetic mutations can change pain sensitivity and behavioral responses to pain. People born genetically insensate to pain-that is, individuals who cannot feel pain-have a mutation in part of a gene that plays a role in cell survival. Using "knockout" animal models-animals genetically engineered to lack a certain gene-scientists are able to visualize how mutations in genes cause animals to become anxious, make noise, rear, freeze, or become hypervigilant. These genetic mutations cause a disruption or alteration in the processing of pain information as it leaves the spinal cord and travels to the brain. Knockout animals can be used to complement efforts aimed at developing new drugs. Plasticity: Following injury, the nervous system undergoes a tremendous reorganization. This phenomenon is known as plasticity. For example, the spinal cord is "rewired" following trauma as nerve cell axons make new contacts, a phenomenon known as "sprouting." This in turn disrupts the cells' supply of trophic factors. Scientists can now identify and study the changes that occur during the processing of pain. For example, using a technique called polymerase chain reaction, abbreviated PCR, scientists can study the genes that are induced by injury and persistent pain. There is evidence that the proteins that are ultimately synthesized by these genes may be targets for new therapies. The dramatic changes that occur with injury and persistent pain underscore that chronic pain should be considered a disease of the nervous system, not just prolonged acute pain or a symptom of an injury. Thus, scientists hope that therapies directed at preventing the long-term changes that occur in the nervous system will prevent the development of chronic pain conditions. Neurotransmitters: Just as mutations in genes may affect behavior, they may also affect a number of neurotransmitters involved in the control of pain. Using sophisticated imaging technologies, investigators can now visualize what is happening chemically in the spinal cord. From this work, new therapies may emerge, therapies that can help reduce or obliterate severe or chronic pain. Hope for the FutureThousands of years ago, ancient peoples attributed pain to spirits and treated it with mysticism and incantations. Over the centuries, science has provided us with a remarkable ability to understand and control pain with medications, surgery, and other treatments. Today, scientists understand a great deal about the causes and mechanisms of pain, and research has produced dramatic improvements in the diagnosis and treatment of a number of painful disorders. For people who fight every day against the limitations imposed by pain, the work of NINDS-supported scientists holds the promise of an even greater understanding of pain in the coming years. Their research offers a powerful weapon in the battle to prolong and improve the lives of people with pain: hope. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
Spine Basics: The Vertebrae, Discs,
and Spinal Cord Stacked on top of one another in the spine are more than 30 bones, the vertebrae, which together form the spine. They are divided into four regions:
The vertebrae are linked by ligaments, tendons, and muscles. Back pain can occur when, for example, someone lifts something too heavy, causing a sprain, pull, strain, or spasm in one of these muscles or ligaments in the back. Between the vertebrae are round, spongy pads of cartilage called discs that act much like shock absorbers. In many cases, degeneration or pressure from overexertion can cause a disc to shift or protrude and bulge, causing pressure on a nerve and resultant pain. When this happens, the condition is called a slipped, bulging, herniated, or ruptured disc, and it sometimes results in permanent nerve damage. The column-like spinal cord is divided into segments similar to the corresponding vertebrae: cervical, thoracic, lumbar, sacral, and coccygeal. The cord also has nerve roots and rootlets which form branch-like appendages leading from its ventral side (that is, the front of the body) and from its dorsal side (that is, the back of the body). Along the dorsal root are the cells of the dorsal root ganglia, which are critical in the transmission of "pain" messages from the cord to the brain. It is here where injury, damage, and trauma become pain. The central nervous system (CNS) refers to the brain and spinal cord together. The peripheral nervous system refers to the cervical, thoracic, lumbar, and sacral nerve trunks leading away from the spine to the limbs. Messages related to function (such as movement) or dysfunction (such as pain) travel from the brain to the spinal cord and from there to other regions in the body and back to the brain again. The autonomic nervous system controls involuntary functions in the body, like perspiration, blood pressure, heart rate, or heart beat. It is divided into the sympathetic and parasympathetic nervous systems. The sympathetic and parasympathetic nervous systems have links to important organs and systems in the body; for example, the sympathetic nervous system controls the heart, blood vessels, and respiratory system, while the parasympathetic nervous system controls our ability to sleep, eat, and digest food. The peripheral nervous system also includes 12 pairs of cranial nerves located on the underside of the brain. Most relay messages of a sensory nature. They include the olfactory (I), optic (II), oculomotor (III), trochlear (IV), trigeminal (V), abducens (VI), facial (VII), vestibulocochlear (VIII), glossopharyngeal (IX), vagus (X), accessory (XI), and hypoglossal (XII) nerves. Neuralgia, as in trigeminal neuralgia, is a term that refers to pain that arises from abnormal activity of a nerve trunk or its branches. The type and severity of pain associated with neuralgia vary widely. Phantom Pain: How Does the Brain
Feel? Sometimes, when a limb is removed during an amputation, an individual will continue to have an internal sense of the lost limb. This phenomenon is known as phantom limb and accounts describing it date back to the 1800s. Similarly, many amputees are frequently aware of severe pain in the absent limb. Their pain is real and is often accompanied by other health problems, such as depression. What causes this phenomenon? Scientists believe that following amputation, nerve cells "rewire" themselves and continue to receive messages, resulting in a remapping of the brain's circuitry. The brain's ability to restructure itself, to change and adapt following injury, is called plasticity (see section on Plasticity). Our understanding of phantom pain has improved tremendously in recent years. Investigators previously believed that brain cells affected by amputation simply died off. They attributed sensations of pain at the site of the amputation to irritation of nerves located near the limb stump. Now, using imaging techniques such as positron emission tomography (PET) and magnetic resonance imaging (MRI), scientists can actually visualize increased activity in the brain's cortex when an individual feels phantom pain. When study participants move the stump of an amputated limb, neurons in the brain remain dynamic and excitable. Surprisingly, the brain's cells can be stimulated by other body parts, often those located closest to the missing limb. Treatments for phantom pain may include analgesics, anticonvulsants, and other types of drugs; nerve blocks; electrical stimulation; psychological counseling, biofeedback, hypnosis, and acupuncture; and, in rare instances, surgery. Chili Peppers, Capsaicin, and Pain The hot feeling, red face, and watery eyes you experience when you bite into a red chili pepper may make you reach for a cold drink, but that reaction has also given scientists important information about pain. The chemical found in chili peppers that causes those feelings is capsaicin (pronounced cap-SAY-sin), and it works its unique magic by grabbing onto receptors scattered along the surface of sensitive nerve cells in the mouth. In 1997, scientists at the University of California at San Francisco discovered a gene for a capsaicin receptor, called the vanilloid receptor. Once in contact with capsaicin, vanilloid receptors open and pain signals are sent from the peripheral nociceptor and through central nervous system circuits to the brain. Investigators have also learned that this receptor plays a role in the burning type of pain commonly associated with heat, such as the kind you experience when you touch your finger to a hot stove. The vanilloid receptor functions as a sort of "ouch gateway," enabling us to detect burning hot pain, whether it originates from a 3-alarm habanera chili or from a stove burner. Capsaicin is currently available as a prescription or over-the-counter cream for the treatment of a number of pain conditions, such as shingles. It works by reducing the amount of substance P found in nerve endings and interferes with the transmission of pain signals to the brain. Individuals can become desensitized to the compound, however, perhaps because of long-term damage to nerve tissue. Some individuals find the burning sensation they experience when using capsaicin cream to be intolerable, especially when they are already suffering from a painful condition, such as postherpetic neuralgia. Soon, however, better treatments that relieve pain by blocking vanilloid receptors may arrive in drugstores. As a painkiller, marijuana or, by its Latin name, cannabis, continues to remain highly controversial. In the eyes of many individuals campaigning on its behalf, marijuana rightfully belongs with other pain remedies. In fact, for many years, it was sold under highly controlled conditions in cigarette form by the Federal government for just that purpose. In 1997, the National Institutes of Health held a workshop to discuss research on the possible therapeutic uses for smoked marijuana. Panel members from a number of fields reviewed published research and heard presentations from pain experts. The panel members concluded that, because there are too few scientific studies to prove marijuana's therapeutic utility for certain conditions, additional research is needed. There is evidence, however, that receptors to which marijuana binds are found in many brain regions that process information that can produce pain. Nerve blocks may involve local anesthesia, regional anesthesia or analgesia, or surgery; dentists routinely use them for traditional dental procedures. Nerve blocks can also be used to prevent or even diagnose pain. In the case of a local nerve block, any one of a number of local anesthetics may be used; the names of these compounds, such as lidocaine or novocaine, usually have an aine ending. Regional blocks affect a larger area of the body. Nerve blocks may also take the form of what is commonly called an epidural, in which a drug is administered into the space between the spine's protective covering (the dura) and the spinal column. This procedure is most well known for its use during childbirth. Morphine and methadone are opioid narcotics (such drugs end in ine or one) that are sometimes used for regional analgesia and are administered as an injection. Neurolytic blocks employ injection of chemical agents such as alcohol, phenol, or glycerol to block pain messages and are most often used to treat cancer pain or to block pain in the cranial nerves (see The Nervous Systems). In some cases, a drug called guanethidine is administered intravenously in order to accomplish the block. Surgical blocks are performed on cranial, peripheral, or sympathetic nerves. They are most often done to relieve the pain of cancer and extreme facial pain, such as that experienced with trigeminal neuralgia. There are several different types of surgical nerve blocks and they are not without problems and complications. Nerve blocks can cause muscle paralysis and, in many cases, result in at least partial numbness. For that reason, the procedure should be reserved for a select group of patients and should only be performed by skilled surgeons. Types of surgical nerve blocks include:
NIH Publication No. 01-2406
|
|
Introduction
What Is Dementia? What Are the Different Kinds of Dementia? Secondary Dementias Dementias in Children What Other Conditions Can Cause Dementia? What Conditions Are Not Dementia? What Causes Dementia? What Are the Risk Factors for Dementia? How Is Dementia Diagnosed? Is There Any Treatment? Can Dementia be Prevented? What Kind of Care Does a Person with Dementia Need? What Research Is Being Done? How Can I Help Research? Where can I get more information? Glossary IntroductionA woman in her early 50s was admitted to a hospital because of increasingly odd behavior. Her family reported that she had been showing memory problems and strong feelings of jealousy. She also had become disoriented at home and was hiding objects. During a doctor's examination, the woman was unable to remember her husband's name, the year, or how long she had been at the hospital. She could read but did not seem to understand what she read, and she stressed the words in an unusual way. She sometimes became agitated and seemed to have hallucinations and irrational fears. This woman, known as Auguste D., was the first person reported to have the disease now known as Alzheimer's disease * (AD) after Alois Alzheimer, the German doctor who first described it. After Auguste D. died in 1906, doctors examined her brain and found that it appeared shrunken and contained several unusual features, including strange clumps of protein called plaques and tangled fibers inside the nerve cells. Memory impairments and other symptoms of dementia, which means "deprived of mind," had been described in older adults since ancient times. However, because Auguste D. began to show symptoms at a relatively early age, doctors did not think her disease could be related to what was then called "senile dementia. "The word senile is derived from a Latin term that means, roughly, "old age." It is now clear that AD is a major cause of dementia in elderly people as well as in relatively young adults. Furthermore, we know that it is only one of many disorders that can lead to dementia. The U. S. Congress Office of Technology Assessment estimates that as many as 6.8 million people in the United States have dementia, and at least 1.8 million of those are severely affected. Studies in some communities have found that almost half of all people age 85 and older have some form of dementia. Although it is common in very elderly individuals, dementia is not a normal part of the aging process. Many people live into their 90s and even 100s without any symptoms of dementia. Besides senile dementia, other terms often used to describe dementia include senility and organic brain syndrome. Senility and senile dementia are outdated terms that reflect the formerly widespread belief that dementia was a normal part of aging. Organic brain syndrome is a general term that refers to physical disorders (not psychiatric in origin) that impair mental functions. Research in the last 30 years has led to a greatly improved
understanding of what dementia is, who gets it, and how it
develops and affects the brain. This work is beginning to
pay off with better diagnostic techniques, improved treatments,
and even potential ways of preventing these diseases. What Is Dementia?Dementia is not a specific disease. It is a descriptive term for a collection of symptoms that can be caused by a number of disorders that affect the brain. People with dementia have significantly impaired intellectual functioning that interferes with normal activities and relationships. They also lose their ability to solve problems and maintain emotional control, and they may experience personality changes and behavioral problems such as agitation, delusions, and hallucinations. While memory loss is a common symptom of dementia, memory loss by itself does not mean that a person has dementia. Doctors diagnose dementia only if two or more brain functions - such as memory, language skills, perception, or cognitive skills including reasoning and judgment - are significantly impaired without loss of consciousness. There are many disorders that can cause dementia. Some, such as AD, lead to a progressive loss of mental functions. But other types of dementia can be halted or reversed with appropriate treatment. With AD and many other types of dementia, disease processes cause many nerve cells to stop functioning, lose connections with other neurons, and die. In contrast, normal aging does not result in the loss of large numbers of neurons in the brain. What Are the Different Kinds of Dementia?Dementing disorders can be classified many different ways. These classification schemes attempt to group disorders that have particular features in common, such as whether they are progressive or what parts of the brain are affected. Some frequently used classifications include the following: · Cortical dementia - dementia where the brain damage primarily affects the brain's cortex, or outer layer. Cortical dementias tend to cause problems with memory, language, thinking, and social behavior. · Subcortical dementia - dementia that affects parts of the brain below the cortex. Subcortical dementia tends to cause changes in emotions and movement in addition to problems with memory. · Progressive dementia - dementia that gets worse over time, gradually interfering with more and more cognitive abilities. · Primary dementia - dementia such as AD that does not result from any other disease. · Secondary dementia - dementia that occurs as a result of a physical disease or injury. Some types of dementia fit into more than one of these classifications. For example, AD is considered both a progressive and a cortical dementia. Alzheimer's disease is the most common cause of dementia in people aged 65 and older. Experts believe that up to 4 million people in the United States are currently living with the disease: one in ten people over the age of 65 and nearly half of those over 85 have AD. At least 360,000 Americans are diagnosed with AD each year and about 50,000 are reported to die from it. In most people, symptoms of AD appear after age 60. However, there are some early-onset forms of the disease, usually linked to a specific gene defect, which may appear as early as age 30. AD usually causes a gradual decline in cognitive abilities, usually during a span of 7 to 10 years. Nearly all brain functions, including memory, movement, language, judgment, behavior, and abstract thinking, are eventually affected. AD is characterized by two abnormalities in the brain: amyloid plaques and neurofibrillary tangles. Amyloid plaques, which are found in the tissue between the nerve cells, are unusual clumps of a protein called beta amyloid along with degenerating bits of neurons and other cells. Neurofibrillary tangles are bundles of twisted filaments found within neurons. These tangles are largely made up of a protein called tau. In healthy neurons, the tau protein helps the functioning of microtubules, which are part of the cell's structural support and deliver substances throughout the nerve cell. However, in AD, tau is changed in a way that causes it to twist into pairs of helical filaments that collect into tangles. When this happens, the microtubules cannot function correctly and they disintegrate. This collapse of the neuron's transport system may impair communication between nerve cells and cause them to die. Researchers do not know if amyloid plaques and neurofibrillary tangles are harmful or if they are merely side effects of the disease process that damages neurons and leads to the symptoms of AD. They do know that plaques and tangles usually increase in the brain as AD progresses. In the early stages of AD, patients may experience memory impairment, lapses of judgment, and subtle changes in personality. As the disorder progresses, memory and language problems worsen and patients begin to have difficulty performing activities of daily living, such as balancing a checkbook or remembering to take medications. They also may have visuospatial problems, such as difficulty navigating an unfamiliar route. They may become disoriented about places and times, may suffer delusions (such as the idea that someone is stealing from them or that their spouse is being unfaithful), and may become short-tempered and hostile. During the late stages of the disease, patients begin to lose the ability to control motor functions. They may have difficulty swallowing and lose bowel and bladder control. They eventually lose the ability to recognize family members and to speak. As AD progresses, it begins to affect the person's emotions and behavior. Most people with AD eventually develop symptoms such as aggression, agitation, depression, sleeplessness, or delusions. On average, patients with AD live for 8 to 10 years after they are diagnosed. However, some people live as long as 20 years. Patients with AD often die of aspiration pneumonia because they lose the ability to swallow late in the course of the disease. Vascular dementia is the second most common cause of dementia, after AD. It accounts for up to 20 percent of all dementias and is caused by brain damage from cerebrovascular or cardiovascular problems - usually strokes. It also may result from genetic diseases, endocarditis (infection of a heart valve), or amyloid angiopathy (a process in which amyloid protein builds up in the brain's blood vessels, sometimes causing hemorrhagic or "bleeding" strokes). In many cases, it may coexist with AD. The incidence of vascular dementia increases with advancing age and is similar in men and women. Symptoms of vascular dementia often begin suddenly, frequently after a stroke. Patients may have a history of high blood pressure, vascular disease, or previous strokes or heart attacks. Vascular dementia may or may not get worse with time, depending on whether the person has additional strokes. In some cases, symptoms may get better with time. When the disease does get worse, it often progresses in a stepwise manner, with sudden changes in ability. Vascular dementia with brain damage to the mid-brain regions, however, may cause a gradual, progressive cognitive impairment that may look much like AD. Unlike people with AD, people with vascular dementia often maintain their personality and normal levels of emotional responsiveness until the later stages of the disease. People with vascular dementia frequently wander at night and often have other problems commonly found in people who have had a stroke, including depression and incontinence. There are several types of vascular dementia, which vary slightly in their causes and symptoms. One type, called multi-infarct dementia (MID), is caused by numerous small strokes in the brain. MID typically includes multiple damaged areas, called infarcts, along with extensive lesions in the white matter, or nerve fibers, of the brain. Because the infarcts in MID affect isolated areas of the brain, the symptoms are often limited to one side of the body or they may affect just one or a few specific functions, such as language. Neurologists call these "local" or "focal" symptoms, as opposed to the "global" symptoms seen in AD, which affect many functions and are not restricted to one side of the body. Although not all strokes cause dementia, in some cases a single stroke can damage the brain enough to cause dementia. This condition is called single-infarct dementia. Dementia is more common when the stroke takes place on the left side (hemisphere) of the brain and/or when it involves the hippocampus, a brain structure important for memory. Another type of vascular dementia is called Binswanger's disease. This rare form of dementia is characterized by damage to small blood vessels in the white matter of the brain (white matter is found in the inner layers of the brain and contains many nerve fibers coated with a whitish, fatty substance called myelin). Binswanger's disease leads to brain lesions, loss of memory, disordered cognition, and mood changes. Patients with this disease often show signs of abnormal blood pressure, stroke, blood abnormalities, disease of the large blood vessels in the neck, and/or disease of the heart valves. Other prominent features include urinary incontinence, difficulty walking, clumsiness, slowness, lack of facial expression, and speech difficulty. These symptoms, which usually begin after the age of 60, are not always present in all patients and may sometimes appear only temporarily. Treatment of Binswanger's disease is symptomatic, and may include the use of medications to control high blood pressure, depression, heart arrhythmias, and low blood pressure. The disorder often includes episodes of partial recovery. Another type of vascular dementia is linked to a rare hereditary disorder called CADASIL, which stands for cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy. CADASIL is linked to abnormalities of a specific gene, Notch3, which is located on chromosome 19. This condition causes multi-infarct dementia as well as stroke, migraine with aura, and mood disorders. The first symptoms usually appear in people who are in their twenties, thirties, or forties and affected individuals often die by age 65. Researchers believe most people with CADASIL go undiagnosed, and the actual prevalence of the disease is not yet known. Other causes of vascular dementia include vasculitis, an inflammation of the blood vessel system; profound hypotension (low blood pressure); and lesions caused by brain hemorrhage. The autoimmune disease lupus erythematosus and the inflammatory disease temporal arteritis can also damage blood vessels in a way that leads to vascular dementia. Lewy body dementia (LBD) is one of the most common types of progressive dementia. LBD usually occurs sporadically, in people with no known family history of the disease. However, rare familial cases have occasionally been reported. In LBD, cells die in the brain's cortex, or outer layer, and in a part of the mid-brain called the substantia nigra. Many of the remaining nerve cells in the substantia nigra contain abnormal structures called Lewy bodies that are the hallmark of the disease. Lewy bodies may also appear in the brain's cortex, or outer layer. Lewy bodies contain a protein called alpha-synuclein that has been linked to Parkinson's disease and several other disorders. Researchers, who sometimes refer to these disorders collectively as "synucleinopathies," do not yet know why this protein accumulates inside nerve cells in LBD. The symptoms of LBD overlap with AD in many ways, and may include memory impairment, poor judgment, and confusion. However, LBD typically also includes visual hallucinations, parkinsonian symptoms such as a shuffling gait and flexed posture, and day-to-day fluctuations in the severity of symptoms. Patients with LBD live an average of 7 years after symptoms begin. There is no cure for LBD, and treatments are aimed at controlling the parkinsonian and psychiatric symptoms of the disorder. Patients sometimes respond dramatically to treatment with antiparkinsonian drugs and/or cholinesterase inhibitors, such as those used for AD. Some studies indicate that neuroleptic drugs, such as clozapine and olanzapine, also can reduce the psychiatric symptoms of this disease. But neuroleptic drugs may cause severe adverse reactions, so other therapies should be tried first and patients using these drugs should be closely monitored. Lewy bodies are often found in the brains of people with Parkinson's and AD. These findings suggest that either LBD is related to these other causes of dementia or that the diseases sometimes coexist in the same person. Frontotemporal dementia (FTD), sometimes called frontal lobe dementia, describes a group of diseases characterized by degeneration of nerve cells - especially those in the frontal and temporal lobes of the brain. Unlike AD, FTD usually does not include formation of amyloid plaques. In many people with FTD, there is an abnormal form of tau protein in the brain, which accumulates into neurofibrillary tangles. This disrupts normal cell activities and may cause the cells to die. Experts believe FTD accounts for 2 to 10 percent of all cases of dementia. Symptoms of FTD usually appear between the ages of 40 and 65. In many cases, people with FTD have a family history of dementia, suggesting that there is a strong genetic factor in the disease. The duration of FTD varies, with some patients declining rapidly over 2 to 3 years and others showing only minimal changes for many years. People with FTD live with the disease for an average of 5 to 10 years after diagnosis. Because structures found in the frontal and temporal lobes of the brain control judgment and social behavior, people with FTD often have problems maintaining normal interactions and following social conventions. They may steal or exhibit impolite and socially inappropriate behavior, and they may neglect their normal responsibilities. Other common symptoms include loss of speech and language, compulsive or repetitive behavior, increased appetite, and motor problems such as stiffness and balance problems. Memory loss also may occur, although it typically appears late in the disease. In one type of FTD called Pick's disease, certain nerve cells become abnormal and swollen before they die. These swollen, or ballooned, neurons are one hallmark of the disease. The brains of people with Pick's disease also have abnormal structures called Pick bodies, composed largely of the protein tau, inside the neurons. The cause of Pick's disease is unknown, but it runs in some families and thus it is probably due at least in part to a faulty gene or genes. The disease usually begins after age 50 and causes changes in personality and behavior that gradually worsen over time. The symptoms of Pick's disease are very similar to those of AD, and may include inappropriate social behavior, loss of mental flexibility, language problems, and difficulty with thinking and concentration. There is currently no way to slow the progressive degeneration found in Pick's disease. However, medication may be helpful in reducing aggression and other behavioral problems, and in treating depression. In some cases, familial FTD is linked to a mutation in the tau gene. This disorder, called frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), is much like other types of FTD but often includes psychiatric symptoms such as delusions and hallucinations. Primary progressive aphasia (PPA) is a type of FTD that may begin in people as early as their forties. "Aphasia" is a general term used to refer to deficits in language functions, such as speaking, understanding what others are saying, and naming common objects. In PPA one or more of these functions can become impaired. Symptoms often begin gradually and progress slowly over a period of years. As the disease progresses, memory and attention may also be impaired and patients may show personality and behavior changes. Many, but not all, people with PPA eventually develop symptoms of dementia. HIV-associated dementia (HAD) results from infection with the human immunodeficiency virus (HIV) that causes AIDS. HAD can cause widespread destruction of the brain's white matter. This leads to a type of dementia that generally includes impaired memory, apathy, social withdrawal, and difficulty concentrating. People with HAD often develop movement problems as well. There is no specific treatment for HAD, but AIDS drugs can delay onset of the disease and may help to reduce symptoms. Huntington's disease (HD) is a hereditary disorder caused by a faulty gene for a protein called huntingtin. The children of people with the disorder have a 50 percent chance of inheriting it. The disease causes degeneration in many regions of the brain and spinal cord. Symptoms of HD usually begin when patients are in their thirties or forties, and the average life expectancy after diagnosis is about 15 years. Cognitive symptoms of HD typically begin with mild personality changes, such as irritability, anxiety, and depression, and progress to severe dementia. Many patients also show psychotic behavior. HD causes chorea - involuntary jerky, arrhythmic movements of the body - as well as muscle weakness, clumsiness, and gait disturbances. Dementia pugilistica, also called chronic traumatic encephalopathy or Boxer's syndrome, is caused by head trauma, such as that experienced by people who have been punched many times in the head during boxing. The most common symptoms of the condition are dementia and parkinsonism, which can appear many years after the trauma ends. Affected individuals may also develop poor coordination and slurred speech. A single traumatic brain injury may also lead to a disorder called post-traumatic dementia (PTD). PTD is much like dementia pugilistica but usually also includes long-term memory problems. Other symptoms vary depending on which part of the brain was damaged by the injury. Corticobasal degeneration (CBD) is a progressive disorder characterized by nerve cell loss and atrophy of multiple areas of the brain. Brain cells from people with CBD often have abnormal accumulations of the protein tau. CBD usually progresses gradually over the course of 6 to 8 years. Initial symptoms, which typically begin at or around age 60, may first appear on one side of the body but eventually will affect both sides. Some of the symptoms, such as poor coordination and rigidity, are similar to those found in Parkinson's disease. Other symptoms may include memory loss, dementia, visual-spatial problems, apraxia (loss of the ability to make familiar, purposeful movements), hesitant and halting speech, myoclonus (involuntary muscular jerks), and dysphagia (difficulty swallowing). Death is often caused by pneumonia or other secondary problems such as sepsis (severe infection of the blood) or pulmonary embolism (a blood clot in the lungs). There are no specific treatments available for CBD. Drugs such as clonazepam may help with myoclonus, however, and occupational, physical, and speech therapy can help in managing the disabilities associated with this disease. The symptoms of the disease often do not respond to Parkinson's medications or other drugs. Creutzfeldt-Jakob disease (CJD) is a rare, degenerative, fatal brain disorder that affects about one in every million people per year worldwide. Symptoms usually begin after age 60 and most patients die within 1 year. Many researchers believe CJD results from an abnormal form of a protein called a prion. Most cases of CJD occur sporadically - that is, in people who have no known risk factors for the disease. However, about 5 to 10 percent of cases of CJD in the United States are hereditary, caused by a mutation in the gene for the prion protein. In rare cases, CJD can also be acquired through exposure to diseased brain or nervous system tissue, usually through certain medical procedures. There is no evidence that CJD is contagious through the air or through casual contact with a CJD patient. Patients with CJD may initially experience problems with muscular coordination; personality changes, including impaired memory, judgment, and thinking; and impaired vision. Other symptoms may include insomnia and depression. As the illness progresses, mental impairment becomes severe. Patients often develop myoclonus and they may go blind. They eventually lose the ability to move and speak, and go into a coma. Pneumonia and other infections often occur in these patients and can lead to death. CJD belongs to a family of human and animal diseases known as the transmissible spongiform encephalopathies (TSEs). Spongiform refers to the characteristic appearance of infected brains, which become filled with holes until they resemble sponges when viewed under a microscope. CJD is the most common of the known human TSEs. Others include fatal familial insomnia and Gerstmann-Straussler-Scheinker disease (see below). In recent years, a new type of CJD, called variant CJD (vCJD), has been found in Great Britain and several other European countries. The initial symptoms of vCJD are different from those of classic CJD and the disorder typically occurs in younger patients. Research suggests that vCJD may have resulted from human consumption of beef from cattle with a TSE disease called bovine spongiform encephalopathy (BSE), also known as "mad cow disease." Other rare hereditary dementias include Gerstmann-Straussler-Scheinker (GSS) disease, fatal familial insomnia, familial British dementia, and familial Danish dementia. Symptoms of GSS typically include ataxia and progressive dementia that begins when people are between 50 and 60 years old. The disease may last for several years before patients eventually die. Fatal familial insomnia causes degeneration of a brain region called the thalamus, which is partially responsible for controlling sleep. It causes a progressive insomnia that eventually leads to a complete inability to sleep. Other symptoms may include poor reflexes, dementia, hallucinations, and eventually coma. It can be fatal within 7 to 13 months after symptoms begin but may last longer. Familial British dementia and familial Danish dementia have been linked to two different defects in a gene found on chromosome 13. The symptoms of both diseases include progressive dementia, paralysis, and loss of balance. Secondary DementiasDementia may occur in patients who have other disorders that primarily affect movement or other functions. These cases are often referred to as secondary dementias. The relationship between these disorders and the primary dementias is not always clear. For instance, people with advanced Parkinson's disease, which is primarily a movement disorder, sometimes develop symptoms of dementia. Many Parkinson's patients also have amyloid plaques and neurofibrillary tangles like those found in AD. The two diseases may be linked in a yet-unknown way, or they may simply coexist in some people. People with Parkinson's and associated dementia sometimes show signs of Lewy body dementia or progressive supranuclear palsy at autopsy, suggesting that these diseases may also overlap with Parkinson's or that Parkinson's is sometimes misdiagnosed. Other disorders that may include symptoms of dementia include
multiple sclerosis; presenile dementia with motor neuron
disease, also called ALS dementia; olivopontocerebellar atrophy
(OPCA); Wilson's disease; and normal pressure hydrocephalus
(NPH).† Dementias in ChildrenWhile it is usually found in adults, dementia can also occur in children. For example, infections and poisoning can lead to dementia in people of any age. In addition, some disorders unique to children can cause dementia. Niemann-Pick disease is a group of inherited disorders that affect metabolism and are caused by specific genetic mutations. Patients with Niemann-Pick disease cannot properly metabolize cholesterol and other lipids. Consequently, excessive amounts of cholesterol accumulate in the liver and spleen and excessive amounts of other lipids accumulate in the brain. Symptoms may include dementia, confusion, and problems with learning and memory. These diseases usually begin in young school-age children but may also appear during the teen years or early adulthood. Batten disease is a fatal, hereditary disorder of the nervous system that begins in childhood. Symptoms are linked to a buildup of substances called lipopigments in the body's tissues. The early symptoms include personality and behavior changes, slow learning, clumsiness, or stumbling. Over time, affected children suffer mental impairment, seizures, and progressive loss of sight and motor skills. Eventually, children with Batten disease develop dementia and become blind and bedridden. The disease is often fatal by the late teens or twenties. Lafora body disease is a rare genetic disease that causes seizures, rapidly progressive dementia, and movement problems. These problems usually begin in late childhood or the early teens. Children with Lafora body disease have microscopic structures called Lafora bodies in the brain, skin, liver, and muscles. Most affected children die within 2 to 10 years after the onset of symptoms. A number of other childhood-onset disorders can include
symptoms of dementia. Among these are mitochondrial myopathies,
Rasmussen's encephalitis, mucopolysaccharidosis III (Sanfilippo
syndrome), neurodegeneration with brain iron accumulation,
and leukodystrophies such as Alexander disease, Schilder's
disease, and metachromatic leukodystrophy. † What Other Conditions Can Cause Dementia?Doctors have identified many other conditions that can cause dementia or dementia-like symptoms. Many of these conditions are reversible with appropriate treatment. Reactions to medications. Medications can sometimes lead to reactions or side effects that mimic dementia. These dementia-like effects can occur in reaction to just one drug or they can result from drug interactions. They may have a rapid onset or they may develop slowly over time. Metabolic problems and endocrine abnormalities. Thyroid problems can lead to apathy, depression, or dementia. Hypoglycemia, a condition in which there is not enough sugar in the bloodstream, can cause confusion or personality changes. Too little or too much sodium or calcium can also trigger mental changes. Some people have an impaired ability to absorb vitamin B12, which creates a condition called pernicious anemia that can cause personality changes, irritability, or depression. Tests can determine if any of these problems are present. Nutritional deficiencies. Deficiencies of thiamine (vitamin B1) frequently result from chronic alcoholism and can seriously impair mental abilities, in particular memories of recent events. Severe deficiency of vitamin B6 can cause a neurological illness called pellagra that may include dementia. Deficiencies of vitamin B12 also have been linked to dementia in some cases. Dehydration can also cause mental impairment that can resemble dementia. Infections. Many infections can cause neurological symptoms, including confusion or delirium, due to fever or other side effects of the body's fight to overcome the infection. Meningitis and encephalitis, which are infections of the brain or the membrane that covers it, can cause confusion, sudden severe dementia, withdrawal from social interaction, impaired judgment, or memory loss. Untreated syphilis also can damage the nervous system and cause dementia. In rare cases, Lyme disease can cause memory or thinking difficulties. People in the advanced stages of AIDS also may develop a form of dementia (see HIV-associated dementia, page 14). People with compromised immune systems, such as those with leukemia and AIDS, may also develop an infection called progressive multifocal leukoencephalopathy (PML). PML is caused by a common human polyomavirus, JC virus, and leads to damage or destruction of the myelin sheath that covers nerve cells. PML can lead to confusion, difficulty with thinking or speaking, and other mental problems. Subdural hematomas. Subdural hematomas, or bleeding between the brain's surface and its outer covering (the dura), can cause dementia-like symptoms and changes in mental function. Poisoning. Exposure to lead, other heavy metals, or other poisonous substances can lead to symptoms of dementia. These symptoms may or may not resolve after treatment, depending on how badly the brain is damaged. People who have abused substances such as alcohol and recreational drugs sometimes display signs of dementia even after the substance abuse has ended. This condition is known as substance-induced persisting dementia. Brain tumors. In rare cases, people with brain tumors may develop dementia because of damage to their brains. Symptoms may include changes in personality, psychotic episodes, or problems with speech, language, thinking, and memory. Anoxia. Anoxia and a related term, hypoxia, are often used interchangeably to describe a state in which there is a diminished supply of oxygen to an organ's tissues. Anoxia may be caused by many different problems, including heart attack, heart surgery, severe asthma, smoke or carbon monoxide inhalation, high-altitude exposure, strangulation, or an overdose of anesthesia. In severe cases of anoxia the patient may be in a stupor or a coma for periods ranging from hours to days, weeks, or months. Recovery depends on the severity of the oxygen deprivation. As recovery proceeds, a variety of psychological and neurological abnormalities, such as dementia or psychosis, may occur. The person also may experience confusion, personality changes, hallucinations, or memory loss. Heart and lung problems. The brain requires a high level of oxygen in order to carry out its normal functions. Therefore, problems such as chronic lung disease or heart problems that prevent the brain from receiving adequate oxygen can starve brain cells and lead to the symptoms of dementia. What Conditions Are Not Dementia?Age-related cognitive decline. As people age, they usually experience slower information processing and mild memory impairment. In addition, their brains frequently decrease in volume and some nerve cells, or neurons, are lost. These changes, called age-related cognitive decline, are normal and are not considered signs of dementia. Mild cognitive impairment. Some people develop cognitive and memory problems that are not severe enough to be diagnosed as dementia but are more pronounced than the cognitive changes associated with normal aging. This condition is called mild cognitive impairment. Although many patients with this condition later develop dementia, some do not. Many researchers are studying mild cognitive impairment to find ways to treat it or prevent it from progressing to dementia. Depression. People with depression are frequently passive or unresponsive, and they may appear slow, confused, or forgetful. Other emotional problems can also cause symptoms that sometimes mimic dementia. Delirium. Delirium is characterized by confusion and rapidly altering mental states. The person may also be disoriented, drowsy, or incoherent, and may exhibit personality changes. Delirium is usually caused by a treatable physical or psychiatric illness, such as poisoning or infections. Patients with delirium often, though not always, make a full recovery after their underlying illness is treated. What Causes Dementia?All forms of dementia result from the death of nerve cells and/or the loss of communication among these cells. The human brain is a very complex and intricate machine and many factors can interfere with its functioning. Researchers have uncovered many of these factors, but they have not yet been able to fit these puzzle pieces together in order to form a complete picture of how dementias develop. Many types of dementia, including AD, Lewy body dementia, Parkinson's dementia, and Pick's disease, are characterized by abnormal structures called inclusions in the brain. Because these inclusions, which contain abnormal proteins, are so common in people with dementia, researchers suspect that they play a role in the development of symptoms. However, that role is unknown, and in some cases the inclusions may simply be a side effect of the disease process that leads to the dementia. Genes clearly play a role in the development of some kinds of dementia. However, in AD and many other disorders, the dementia usually cannot be tied to a single abnormal gene. Instead, these forms of dementia appear to result from a complex interaction of genes, lifestyle factors, and other environmental influences. Researchers have identified several genes that influence susceptibility to AD. Mutations in three of the known genes for AD - genes that control the production of proteins such as amyloid precursor protein (APP), presenilin 1, and presenilin 2 - are linked to early-onset forms of the disease. Variations in another gene, called apolipoprotein E (apoE), have been linked to an increased risk of late-onset AD. The apoE gene does not cause the disease by itself, but one version of the gene, called apoE epsilon4 (apoE E4), appears to increase the risk of AD. People with two copies of the apoE E4 gene have about ten times the risk of developing AD compared to people without apoE E4. This gene variant seems to encourage amyloid deposition in the brain. One study also found that this gene is associated with shorter survival in men with AD. In contrast, another version of the apoE gene, called apoE E2, appears to protect against AD. Studies have suggested that mutations in another gene, called CYP46, may contribute to an increased risk of developing late-onset sporadic AD. This gene normally produces a protein that helps the brain metabolize cholesterol. Scientists are trying to determine how beta amyloid influences the development of AD. A number of studies indicate that the buildup of this protein initiates a complex chain of events that culminates in dementia. One study found that beta amyloid buildup in the brain triggers cells called microglia, which act like janitors that mop up potentially harmful substances in the brain, to release a potent neurotoxin called peroxynitrite. This may contribute to nerve cell death in AD. Another study found that beta amyloid causes a protein called p35 to be split into two proteins. One of the resulting proteins triggers changes in the tau protein that lead to formation of neurofibrillary tangles. A third study found that beta amyloid activates cell-death enzymes called caspases that alter the tau protein in a way that causes it to form tangles. Researchers believe these tangles may contribute to the neuron death in AD. Vascular dementia can be caused by cerebrovascular disease or any other condition that prevents normal blood flow to the brain. Without a normal supply of blood, brain cells cannot obtain the oxygen they need to work correctly, and they often become so deprived that they die. The causes of other types of dementias vary. Some, such as CJD and GSS, have been tied to abnormal forms of specific proteins. Others, including Huntington's disease and FTDP-17, have been linked to defects in a single gene. Post-traumatic dementia is directly related to brain cell death after injury. HIV-associated dementia is clearly tied to infection by the HIV virus, although the exact way the virus causes damage is not yet certain. For other dementias, such as corticobasal degeneration and most types of frontotemporal dementia, the underlying causes have not yet been identified. What Are the Risk Factors for Dementia?Researchers have identified several risk factors that affect the likelihood of developing one or more kinds of dementia. Some of these factors are modifiable, while others are not. Age. The risk of AD, vascular dementia, and several other dementias goes up significantly with advancing age. Genetics/family history. As described in the section "What Causes Dementia?" researchers have discovered a number of genes that increase the risk of developing AD. Although people with a family history of AD are generally considered to be at heightened risk of developing the disease themselves, many people with a family history never develop the disease, and many without a family history of the disease do get it. In most cases, it is still impossible to predict a specific person's risk of the disorder based on family history alone. Some families with CJD, GSS, or fatal familial insomnia have mutations in the prion protein gene, although these disorders can also occur in people without the gene mutation. Individuals with these mutations are at significantly higher risk of developing these forms of dementia. Abnormal genes are also clearly implicated as risk factors in Huntington's disease, FTDP-17, and several other kinds of dementia. These dementias are described in the section "What are the different kinds of dementia?" Smoking and alcohol use. Several recent studies have found that smoking significantly increases the risk of mental decline and dementia. People who smoke have a higher risk of atherosclerosis and other types of vascular disease, which may be the underlying causes for the increased dementia risk. Studies also have found that drinking large amounts of alcohol appears to increase the risk of dementia. However, other studies have suggested that people who drink moderately have a lower risk of dementia than either those who drink heavily or those who completely abstain from drinking. Atherosclerosis. Atherosclerosis is the buildup of plaque - deposits of fatty substances, cholesterol, and other matter - in the inner lining of an artery. Atherosclerosis is a significant risk factor for vascular dementia, because it interferes with the delivery of blood to the brain and can lead to stroke. Studies have also found a possible link between atherosclerosis and AD. Cholesterol. High levels of low-density lipoprotein (LDL), the so-called bad form of cholesterol, appear to significantly increase a person's risk of developing vascular dementia. Some research has also linked high cholesterol to an increased risk of AD. Plasma homocysteine. Research has shown that a higher-than-average blood level of homocysteine - a type of amino acid - is a strong risk factor for the development of AD and vascular dementia. Diabetes. Diabetes is a risk factor for both AD and vascular dementia. It is also a known risk factor for atherosclerosis and stroke, both of which contribute to vascular dementia. Mild cognitive impairment. While not all people with mild cognitive impairment develop dementia, people with this condition do have a significantly increased risk of dementia compared to the rest of the population. One study found that approximately 40 percent of people over age 65 who were diagnosed with mild cognitive impairment developed dementia within 3 years. Down syndrome. Studies have found that most people with Down syndrome develop characteristic AD plaques and neurofibrillary tangles by the time they reach middle age. Many, but not all, of these individuals also develop symptoms of dementia. How Is Dementia Diagnosed?Doctors employ a number of strategies to diagnose dementia. It is important that they rule out any treatable conditions, such as depression, normal pressure hydrocephalus, or vitamin B12 deficiency, which can cause similar symptoms. Early, accurate diagnosis of dementia is important for patients and their families because it allows early treatment of symptoms. For people with AD or other progressive dementias, early diagnosis may allow them to plan for the future while they can still help to make decisions. These people also may benefit from drug treatment. The "gold standard" for diagnosing dementia, autopsy, does not help the patient or caregivers. Therefore, doctors have devised a number of techniques to help identify dementia with reasonable accuracy while the patient is still alive. Patient history Physical examination Neurological evaluations Cognitive and neuropsychological tests Doctors often use a test called the Mini-Mental State Examination (MMSE) to assess cognitive skills in people with suspected dementia. This test examines orientation, memory, and attention, as well as the ability to name objects, follow verbal and written commands, write a sentence spontaneously, and copy a complex shape. Doctors also use a variety of other tests and rating scales to identify specific types of cognitive problems and abilities. Brain scans The most common types of brain scans are computed tomographic (CT) scans and magnetic resonance imaging (MRI). Doctors frequently request a CT scan of the brain when they are examining a patient with suspected dementia. These scans, which use X-rays to detect brain structures, can show evidence of brain atrophy, strokes and transient ischemic attacks (TIAs), changes to the blood vessels, and other problems such as hydrocephalus and subdural hematomas. MRI scans use magnetic fields and focused radio waves to detect hydrogen atoms in tissues within the body. They can detect the same problems as CT scans but they are better for identifying certain conditions, such as brain atrophy and damage from small TIAs. Doctors also may use electroencephalograms (EEGs) in people with suspected dementia. In an EEG, electrodes are placed on the scalp over several parts of the brain in order to detect and record patterns of electrical activity and check for abnormalities. This electrical activity can indicate cognitive dysfunction in part or all of the brain. Many patients with moderately severe to severe AD have abnormal EEGs. An EEG may also be used to detect seizures, which occur in about 10 percent of AD patients as well as in many other disorders. EEGs also can help diagnose CJD. Several other types of brain scans allow researchers to watch the brain as it functions. These scans, called functional brain imaging, are not often used as diagnostic tools, but they are important in research and they may ultimately help identify people with dementia earlier than is currently possible. Functional brain scans include functional MRI (fMRI), single photon-emission computed tomography (SPECT), positron emission tomography (PET), and magnetoencephalography (MEG). fMRI uses radio waves and a strong magnetic field to measure the metabolic changes that take place in active parts of the brain. SPECT shows the distribution of blood in the brain, which generally increases with brain activity. PET scans can detect changes in glucose metabolism, oxygen metabolism, and blood flow, all of which can reveal abnormalities of brain function. MEG shows the electromagnetic fields produced by the brain's neuronal activity. Laboratory tests Psychiatric evaluation Presymptomatic testing Researchers are examining whether a series of simple cognitive tests, such as matching words with pictures, can predict who will develop dementia. One study suggested that a combination of a verbal learning test and an odor-identification test can help identify AD before symptoms become obvious. Other studies are looking at whether memory tests and brain scans can be useful indicators of future dementia. Is There Any Treatment?While treatments to reverse or halt disease progression are not available for most of the dementias, patients can benefit to some extent from treatment with available medications and other measures, such as cognitive training. Drugs to specifically treat AD and some other progressive dementias are now available and are prescribed for many patients. Although these drugs do not halt the disease or reverse existing brain damage, they can improve symptoms and slow the progression of the disease. This may improve the patient's quality of life, ease the burden on caregivers, and/or delay admission to a nursing home. Many researchers are also examining whether these drugs may be useful for treating other types of dementia. Many people with dementia, particularly those in the early stages, may benefit from practicing tasks designed to improve performance in specific aspects of cognitive functioning. For example, people can sometimes be taught to use memory aids, such as mnemonics, computerized recall devices, or note taking. Behavior modification - rewarding appropriate or positive behavior and ignoring inappropriate behavior - also may help control unacceptable or dangerous behaviors. Alzheimer's disease A fifth drug, memantine (Namenda), is also approved for use in the United States. Unlike other drugs for AD, which affect acetylcholine levels, memantine works by regulating the activity of a neurotransmitter called glutamate that plays a role in learning and memory. Glutamate activity is often disrupted in AD. Because this drug works differently from cholinesterase inhibitors, combining memantine with other AD drugs may be more effective than any single therapy. One controlled clinical trial found that patients receiving donepezil plus memantine had better cognition and other functions than patients receiving donepezil alone. Doctors may also prescribe other drugs, such as anticonvulsants, sedatives, and antidepressants, to treat seizures, depression, agitation, sleep disorders, and other specific problems that can be associated with dementia. In 2005, research showed that use of "atypical" antipsychotic drugs such as olanzapine and risperdone to treat behavioral problems in elderly people with dementia was associated with an elevated risk of death in these patients. Most of the deaths were caused by heart problems or infections. The FDA has issued a public health advisory to alert patients and their caregivers to this safety issue. Vascular dementia The progression of vascular dementia can often be slowed significantly or halted if the underlying vascular risk factors for the disease are treated. To prevent strokes and TIAs, doctors may prescribe medicines to control high blood pressure, high cholesterol, heart disease, and diabetes. Doctors also sometimes prescribe aspirin, warfarin, or other drugs to prevent clots from forming in small blood vessels. When patients have blockages in blood vessels, doctors may recommend surgical procedures, such as carotid endarterectomy, stenting, or angioplasty, to restore the normal blood supply. Medications to relieve restlessness or depression or to help patients sleep better may also be prescribed. Other dementias At present, no medications are approved specifically to treat or prevent FTD and most other types of progressive dementia. However, sedatives, antidepressants, and other medications may be useful in treating specific symptoms and behavioral problems associated with these diseases. Scientists continue to search for specific treatments to help people with Lewy body dementia. Current treatment is symptomatic, often involving the use of medication to control the parkinsonian and psychiatric symptoms. Although antiparkinsonian medication may help reduce tremor and loss of muscle movement, it may worsen symptoms such as hallucinations and delusions. Also, drugs prescribed for psychiatric symptoms may make the movement problems worse. Several studies have suggested that cholinesterase inhibitors may be able to improve cognitive function and behavioral symptoms in patients with Lewy body disease. There is no known treatment that can cure or control CJD. Current treatment is aimed at alleviating symptoms and making the patient as comfortable as possible. Opiate drugs can help relieve pain, and the drugs clonazepam and sodium valproate may help relieve myoclonus. During later stages of the disease, treatment focuses on supportive care, such as administering intravenous fluids and changing the person's position frequently to prevent bedsores. Can Dementia be Prevented?Research has revealed a number of factors that may be able to prevent or delay the onset of dementia in some people. For example, studies have shown that people who maintain tight control over their glucose levels tend to score better on tests of cognitive function than those with poorly controlled diabetes. Several studies also have suggested that people who engage in intellectually stimulating activities, such as social interactions, chess, crossword puzzles, and playing a musical instrument, significantly lower their risk of developing AD and other forms of dementia. Scientists believe mental activities may stimulate the brain in a way that increases the person's "cognitive reserve" - the ability to cope with or compensate for the pathologic changes associated with dementia. Researchers are studying other steps people can take that may help prevent AD in some cases. So far, none of these factors has been definitively proven to make a difference in the risk of developing the disease. Moreover, most of the studies addressed only AD, and the results may or may not apply to other forms of dementia. Nevertheless, scientists are encouraged by the results of these early studies and many believe it will eventually become possible to prevent some forms of dementia. Possible preventive actions include:
The risk of vascular dementia is strongly correlated with risk factors for stroke, including high blood pressure, diabetes, elevated cholesterol levels, and smoking. This type of dementia may be prevented in many cases by changing lifestyle factors, such as excessive weight and high blood pressure, which are associated with an increased risk of cerebrovascular disease. One European study found that treating isolated systolic hypertension (high blood pressure in which only the systolic or top number is high) in people age 60 and older reduced the risk of dementia by 50 percent. These studies strongly suggest that effective use of current treatments can prevent many future cases of vascular dementia. A study published in 2005 found that people with mild cognitive impairment who took 10 mg/day of the drug donepezil had a significantly reduced risk of developing AD during the first two years of treatment, compared to people who received vitamin E or a placebo. By the end of the third year, however, the rate of AD was just as high in the people treated with donepezil as it was in the other two groups. What Kind of Care Does a Person with Dementia Need?People with moderate and advanced dementia typically need round-the-clock care and supervision to prevent them from harming themselves or others. They also may need assistance with daily activities such as eating, bathing, and dressing. Meeting these needs takes patience, understanding, and careful thought by the person's caregivers. A typical home environment can present many dangers and obstacles to a person with dementia, but simple changes can overcome many of these problems. For example, sharp knives, dangerous chemicals, tools, and other hazards should be removed or locked away. Other safety measures include installing bed and bathroom safety rails, removing locks from bedroom and bathroom doors, and lowering the hot water temperature to 120°F (48. 9°C) or less to reduce the risk of accidental scalding. People with dementia also should wear some form of identification at all times in case they wander away or become lost. Caregivers can help prevent unsupervised wandering by adding locks or alarms to outside doors. People with dementia often develop behavior problems because of frustration with specific situations. Understanding and modifying or preventing the situations that trigger these behaviors may help to make life more pleasant for the person with dementia as well as his or her caregivers. For instance, the person may be confused or frustrated by the level of activity or noise in the surrounding environment. Reducing unnecessary activity and noise (such as limiting the number of visitors and turning off the television when it's not in use) may make it easier for the person to understand requests and perform simple tasks. Confusion also may be reduced by simplifying home decorations, removing clutter, keeping familiar objects nearby, and following a predictable routine throughout the day. Calendars and clocks also may help patients orient themselves. People with dementia should be encouraged to continue their normal leisure activities as long as they are safe and do not cause frustration. Activities such as crafts, games, and music can provide important mental stimulation and improve mood. Some studies have suggested that participating in exercise and intellectually stimulating activities may slow the decline of cognitive function in some people. Many studies have found that driving is unsafe for people with dementia. They often get lost and they may have problems remembering or following rules of the road. They also may have difficulty processing information quickly and dealing with unexpected circumstances. Even a second of confusion while driving can lead to an accident. Driving with impaired cognitive functions can also endanger others. Some experts have suggested that regular screening for changes in cognition might help to reduce the number of driving accidents among elderly people, and some states now require that doctors report people with AD to their state motor vehicle department. However, in many cases, it is up to the person's family and friends to ensure that the person does not drive. The emotional and physical burden of caring for someone with dementia can be overwhelming. Support groups can often help caregivers deal with these demands and they can also offer helpful information about the disease and its treatment. It is important that caregivers occasionally have time off from round-the-clock nursing demands. Some communities provide respite facilities or adult day care centers that will care for dementia patients for a period of time, giving the primary caregivers a break. Eventually, many patients with dementia require the services of a full-time nursing home. A list of caregiver organizations and support groups is included at the end of this booklet. What Research Is Being Done?Current research focuses on many different aspects of dementia. This research promises to improve the lives of people affected by the dementias and may eventually lead to ways of preventing or curing these disorders. Causes and prevention Since many dementias and other neurodegenerative diseases have been linked to abnormal clumps of proteins in cells, researchers are trying to learn how these clumps develop, how they affect cells, and how the clumping can be prevented. Some studies are examining whether changes in white matter - nerve fibers lined with myelin - may play a role in the onset of AD. Myelin may erode in AD patients before other changes occur. This may be due to a problem with oligodendrocytes, the cells that produce myelin. Researchers are searching for additional genes that may contribute to AD, and they have identified a number of gene regions that may be involved. Some researchers suggest that people will eventually be screened for a number of genes that contribute to AD and that they will be able to receive treatments that specifically address their individual genetic risks. However, such individualized screening and treatment is still years away. Insulin resistance is common in people with AD, but it is not clear whether the insulin resistance contributes to the development of the disease or if it is merely a side effect. Several studies have found a reduced risk of dementia in people who take cholesterol-lowering drugs called statins. However, it is not yet clear if the apparent effect is due to the drugs or to other factors. Early studies of estrogen suggested that it might help prevent AD in older women. However, a clinical study of several thousand postmenopausal women aged 65 or older found that combination therapy with estrogen and progestin substantially increased the risk of AD. Estrogen alone also appeared to slightly increase the risk of dementia in this study. A 2003 study found that people with HIV-associated dementia have different levels of activity for more than 30 different proteins, compared to people who have HIV but no signs of dementia. The study suggests a possible way to screen HIV patients for the first signs of cognitive impairment, and it may lead to ways of intervening to prevent this form of dementia. Diagnosis Some researchers are investigating whether three-dimensional computer models of PET and MRI images can identify brain changes typical of early AD, before any symptoms appear. This research may lead to ways of preventing the symptoms of the disease. One study found that levels of beta amyloid and tau in spinal fluid can be used to diagnose AD with a sensitivity of 92 percent. If other studies confirm the validity of this test, it may allow doctors to identify people who are beginning to develop the disorder before they start to show symptoms. This would allow treatment at very early stages of the disorder, and may help in testing new treatments to prevent or delay symptoms of the disease. Other researchers have identified factors in the skin and blood of AD patients that are different from those in healthy people. They are trying to determine if these factors can be used to diagnose the disease. Treatment Researchers are also investigating possible methods of gene therapy for AD. In one case, researchers used cells genetically engineered to produce nerve growth factor and transplanted them into monkeys' forebrains. The transplanted cells boosted the amount of nerve growth factors in the brain and seemed to prevent degeneration of acetylcholine-producing neurons in the animals. This suggests that gene therapy might help to reduce or delay symptoms of the disease. Researchers are now testing a similar therapy in a small number of patients. Other researchers have experimented with gene therapy that adds a gene called neprilysin in a mouse model that produces human beta amyloid. They found that increasing the level of neprilysin greatly reduced the amount of beta amyloid in the mice and halted the amyloid-related brain degeneration. They are now trying to determine whether neprilysin gene therapy can improve cognition in mice. A clinical trial called the Vitamins to Slow Alzheimer's Disease (VITAL) study is testing whether high doses of three common B vitamins - folic acid, B12, and B6 - can reduce homocysteine levels and slow the rate of cognitive decline in AD. Since many studies have found evidence of brain inflammation in AD, some researchers have proposed that drugs that control inflammation, such as NSAIDs, might prevent the disease or slow its progression. Studies in mice have suggested that these drugs can limit production of amyloid plaques in the brain. Early studies of these drugs in humans have shown promising results. However, a large NIH-funded clinical trial of two NSAIDS (naproxen and celecoxib) to prevent AD was stopped in late 2004 because of an increase in stroke and heart attack in people taking naproxen, and an unrelated study that linked celecoxib to an increased risk of heart attack. Some studies have suggested that two drugs, pentoxifylline and propentofylline, may be useful in treating vascular dementia. Pentoxifylline improves blood flow, while propentofylline appears to interfere with some of the processes that cause cell death in the brain. One study is testing the safety and effectiveness of donepezil (Aricept) for treating mild dementia in patients with Parkinson's dementia, while another is investigating whether skin patches with the drug selegiline can improve mental function in patients with cognitive problems related to HIV. How Can I Help Research?People with dementia and others who wish to help research on dementing disorders may be able to do so by participating in clinical studies designed to learn more about the disorders or to test potential new therapies. Information about many such studies is available free of charge from the Federal government's database of clinical trials, clinicaltrials.gov (http://clinicaltrials.gov). Information about clinical trials specific to AD is available from the Alzheimer's Disease Clinical Trials Database (www.alzheimers.org/trials), a joint project of the U. S. Food and Drug Administration and the National Institute on Aging (NIA) that is maintained by the NIA's Alzheimer's Disease Education and Referral Center. For clinical trials taking place at the National Institutes of Health, additional information is available from the following office: Patient Recruitment and Public Liaison Office Voluntary health organizations, such as those listed in Information Resources, may be able to provide information about additional clinical studies. Another important way that people can help dementia research is by arranging to donate their brains to brain and tissue banks after they die. Tissue from these banks is made available to qualified researchers so that they can continue their studies of how these diseases develop and how they affect the brain. Brain banks accepting donations include the following: National Disease Research Interchange Human Brain and Spinal Fluid Resource Center UM/NPF Brain Endowment Bank Harvard Brain Tissue Resource Center People who have more than one family member affected by AD also may be able to help research by contributing blood samples to a gene bank. A large initiative to collect such samples was announced in 2003. This large gene bank should accelerate research efforts to identify genes that play a role in AD. People interested in participating in this gene bank can learn more about it at the address and telephone numbers below: Alzheimer's Disease Genetics Initiative Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
acetylcholine - a neurotransmitter that is important for the formation of memories. Studies have shown that levels of acetylcholine are reduced in the brains of people with Alzheimer's disease. Alzheimer's disease - the most common cause of dementia in people aged 65 and older. Nearly all brain functions, including memory, movement, language, judgment, behavior, and abstract thinking, are eventually affected. amyloid plaques - unusual clumps of material found in the tissue between nerve cells. Amyloid plaques, which consist of a protein called beta amyloid along with degenerating bits of neurons and other cells, are a hallmark of Alzheimer's disease. amyloid precursor protein - a normal brain protein that is a precursor for beta amyloid, the abnormal substance found in the characteristic amyloid plaques of Alzheimer's disease patients. apolipoprotein E - a gene that has been linked to an increased risk of Alzheimer's disease. People with a variant form of the gene, called apoE epsilon 4, have about ten times the risk of developing Alzheimer's disease. ataxia - a loss of muscle control. atherosclerosis - a blood vessel disease characterized by the buildup of plaque, or deposits of fatty substances and other matter in the inner lining of an artery. beta amyloid - a protein found in the characteristic clumps of tissue (called plaques) that appear in the brains of Alzheimer's patients. Binswanger's disease - a rare form of dementia characterized by damage to small blood vessels in the white matter of the brain. This damage leads to brain lesions, loss of memory, disordered cognition, and mood changes. CADASIL - a rare hereditary disorder which is linked to a type of vascular dementia. It stands for cerebral autosomal dominant arteriopathy with subcortical infarct and leukoencephalopathy. cholinesterase inhibitors - drugs that slow the breakdown of the neurotransmitter acetylcholine. cognitive training - a type of training in which patients practice tasks designed to improve mental performance. Examples include memory aids, such as mnemonics, and computerized recall devices. computed tomographic (CT) scans - a type of brain scan that uses X-rays to detect brain structures. cortical atrophy - degeneration of the brain's cortex (outer layer). Cortical atrophy is common in many forms of dementia and may be visible on a brain scan. cortical dementia - a type of dementia in which the damage primarily occurs in the brain's cortex, or outer layer. corticobasal degeneration - a progressive disorder characterized by nerve cell loss and atrophy in multiple areas of the brain. Creutzfeldt-Jakob disease - a rare, degenerative, fatal brain disorder believed to be linked to an abnormal form of a protein called a prion. dementia -a term for a collection of symptoms that significantly impair thinking and normal activities and relationships. dementia pugilistica - a form of dementia caused by head trauma such as that experienced by boxers. It is also called chronic traumatic encephalopathy or Boxer's syndrome. electroencephalogram (EEG) - a medical procedure that records patterns of electrical activity in the brain. fatal familial insomnia - an inherited disease that affects a brain region called the thalamus, which is partially responsible for controlling sleep. The disease causes dementia and a progressive insomnia that eventually leads to a complete lack of sleep. frontotemporal dementias - a group of dementias characterized by degeneration of nerve cells, especially those in the frontal and temporal lobes of the brain. FTDP-17 - one of the frontotemporal dementias, linked to a mutation in the tau gene. It is much like other types of the frontotemporal dementias but often includes psychiatric symptoms such as delusions and hallucinations. Gerstmann-Straussler-Scheinker disease - a rare, fatal hereditary disease that causes ataxia and progressive dementia. HIV-associated dementia - a dementia that results from infection with the human immunodeficiency virus (HIV) that causes AIDS. It can cause widespread destruction of the brain's white matter. Huntington's disease - a degenerative hereditary disorder caused by a faulty gene for a protein called huntington. The disease causes degeneration in many regions of the brain and spinal cord and patients eventually develop severe dementia. Lewy body dementia - one of the most common types of progressive dementia, characterized by the presence of abnormal structures called Lewy bodies in the brain. In many ways the symptoms of this disease overlap with those of Alzheimer's disease. magnetic resonance imaging (MRI) - a diagnostic imaging technique that uses magnetic fields and radio waves to produce detailed images of body structures. mild cognitive impairment - a condition associated with impairments in understanding and memory not severe enough to be diagnosed as dementia, but more pronounced than those associated with normal aging. Mini-Mental State Examination - a test used to assess cognitive skills in people with suspected dementia. The test examines orientation, memory, and attention, as well as the ability to name objects, follow verbal and written commands, write a sentence spontaneously, and copy a complex shape. multi-infarct dementia - a type of vascular dementia caused by numerous small strokes in the brain. myelin - a fatty substance that coats and insulates nerve cells. neurofibrillary tangles - bundles of twisted filaments found within neurons, and a characteristic feature found in the brains of Alzheimer's patients. These tangles are largely made up of a protein called tau. neurotransmitter - a type of chemical, such as acetylcholine, that transmits signals from one neuron to another. People with Alzheimer's disease have reduced supplies of acetylcholine. organic brain syndrome - a term that refers to physical disorders (not psychiatric in origin) that impair mental functions. Parkinson's dementia - a secondary dementia that sometimes occurs in people with advanced Parkinson's disease, which is primarily a movement disorder. Many Parkinson's patients have the characteristic amyloid plaques and neurofibrillary tangles found in Alzheimer's disease, but it is not yet clear if the diseases are linked. Pick's disease - a type of frontotemporal dementia where certain nerve cells become abnormal and swollen before they die. The brains of people with Pick's disease have abnormal structures, called Pick bodies, inside the neurons. The symptoms are very similar to those of Alzheimer's diease. plaques - unusual clumps of material found between the tissues of the brain in Alzheimer's disease. See also amyloid plaques. post-traumatic dementia - a dementia brought on by a single traumatic brain injury. It is much like dementia pugilistica, but usually also includes long-term memory problems. presenilin 1 and 2 - proteins produced by genes that influence susceptibility to early-onset Alzheimer's disease. primary dementia - a dementia, such as Alzheimer's disease, that is not the result of another disease. primary progressive aphasia - a type of frontotemporal dementia resulting in deficits in language functions. Many, but not all, people with this type of aphasia eventually develop symptoms of dementia. progressive dementia - a dementia that gets worse over time, gradually interfering with more and more cognitive abilities. secondary dementia - a dementia that occurs as a consequence of another disease or an injury. senile dementia - an outdated term that reflects the formerly widespread belief that dementia was a normal part of aging. The word senile is derived from a Latin term that means, roughly, "old age. " subcortical dementia - dementia that affects parts of the brain below the outer brain layer, or cortex. substance-induced persisting dementia - dementia caused by abuse of substances such as alcohol and recreational drugs that persists even after the substance abuse has ended. tau protein - a protein that helps the functioning of microtubules, which are part of the cell's structural support and help to deliver substances throughout the cell. In Alzheimer's disease, tau is changed in a way that causes it to twist into pairs of helical filaments that collect into tangles. transmissible spongiform encephalopathies - part of a family of human and animal diseases in which brains become filled with holes resembling sponges when examined under a microscope. CJD is the most common of the known transmissible spongiform encephalopathies. vascular dementia - a type of dementia caused by brain damage from cerebrovascular or cardiovascular problems - usually strokes. It accounts for up to 20 percent of all dementias. |
|
Introduction
What Causes Huntington's Disease? How is HD Inherited? What are the Major Effects of the Disease? At What Age Does HD Appear? How is HD Diagnosed? What is Presymptomatic Testing? How is the Presymptomatic Test Conducted? How Does a Person Decide Whether to be Tested? Is There a Treatment for HD? What Kind of Care Does the Individual with HD Need? What Community Resources are Available? What Research is Being Done? Molecular Genetics The HD Gene and Its Product Cell Death in HD Animal Models of HD Fetal Tissue Research Clinical Studies Imaging How Can I Help? What is the Role of Voluntary Organizations? Where can I get more information? Glossary IntroductionIn 1872, the American physician George Huntington wrote about an illness that he called "an heirloom from generations away back in the dim past." He was not the first to describe the disorder, which has been traced back to the Middle Ages at least. One of its earliest names was chorea,* which, as in "choreography," is the Greek word for dance. The term chorea describes how people affected with the disorder writhe, twist, and turn in a constant, uncontrollable dance-like motion. Later, other descriptive names evolved. "Hereditary chorea" emphasizes how the disease is passed from parent to child. "Chronic progressive chorea" stresses how symptoms of the disease worsen over time. Today, physicians commonly use the simple term Huntington's disease (HD) to describe this highly complex disorder that causes untold suffering for thousands of families. More than 15,000 Americans have HD. At least 150,000 others have a 50 percent risk of developing the disease and thousands more of their relatives live with the possibility that they, too, might develop HD. Until recently, scientists understood very little about HD and could only watch as the disease continued to pass from generation to generation. Families saw the disease destroy their loved ones' ability to feel, think, and move. In the last several years, scientists working with support from the National Institute of Neurological Disorders and Stroke (NINDS) have made several breakthroughs in the area of HD research. With these advances, our understanding of the disease continues to improve. This brochure presents information about HD, and about current research progress, to health professionals, scientists, caregivers, and, most important, to those already too familiar with the disorder: the many families who are affected by HD. What Causes Huntington's Disease?HD results from genetically programmed degeneration of nerve cells, called neurons,* in certain areas of the brain. This degeneration causes uncontrolled movements, loss of intellectual faculties, and emotional disturbance. Specifically affected are cells of the basal ganglia, structures deep within the brain that have many important functions, including coordinating movement. Within the basal ganglia, HD especially targets neurons of the striatum, particularly those in the caudate nuclei and the pallidum. Also affected is the brain's outer surface, or cortex, which controls thought, perception, and memory. How is HD Inherited?HD is found in every country of the world. It is a familial disease, passed from parent to child through a mutation or misspelling in the normal gene. A single abnormal gene, the basic biological unit of heredity, produces HD. Genes are composed of deoxyribonucleic acid (DNA), a molecule shaped like a spiral ladder. Each rung of this ladder is composed of two paired chemicals called bases. There are four types of bases—adenine, thymine, cytosine, and guanine—each abbreviated by the first letter of its name: A, T, C, and G. Certain bases always "pair" together, and different combinations of base pairs join to form coded messages. A gene is a long string of this DNA in various combinations of A, T, C, and G. These unique combinations determine the gene's function, much like letters join together to form words. Each person has about 30,000 genes—a billion base pairs of DNA or bits of information repeated in the nuclei of human cells—which determine individual characteristics or traits. Genes are arranged in precise locations along 23 rod-like pairs of chromosomes. One chromosome from each pair comes from an individual's mother, the other from the father. Each half of a chromosome pair is similar to the other, except for one pair, which determines the sex of the individual. This pair has two X chromosomes in females and one X and one Y chromosome in males. The gene that produces HD lies on chromosome 4, one of the 22 non-sex-linked, or "autosomal," pairs of chromosomes, placing men and women at equal risk of acquiring the disease. The impact of a gene depends partly on whether it is dominant or recessive. If a gene is dominant, then only one of the paired chromosomes is required to produce its called-for effect. If the gene is recessive, both parents must provide chromosomal copies for the trait to be present. HD is called an autosomal dominant disorder because only one copy of the defective gene, inherited from one parent, is necessary to produce the disease. The genetic defect responsible for HD is a small sequence of DNA on chromosome 4 in which several base pairs are repeated many, many times. The normal gene has three DNA bases, composed of the sequence CAG. In people with HD, the sequence abnormally repeats itself dozens of times. Over time—and with each successive generation—the number of CAG repeats may expand further. Each parent has two copies of every chromosome but gives only one copy to each child. Each child of an HD parent has a 50-50 chance of inheriting the HD gene. If a child does not inherit the HD gene, he or she will not develop the disease and cannot pass it to subsequent generations. A person who inherits the HD gene, and survives long enough, will sooner or later develop the disease. In some families, all the children may inherit the HD gene; in others, none do. Whether one child inherits the gene has no bearing on whether others will or will not share the same fate. A small number of cases of HD are sporadic, that is, they occur even though there is no family history of the disorder. These cases are thought to be caused by a new genetic mutation-an alteration in the gene that occurs during sperm development and that brings the number of CAG repeats into the range that causes disease. What are the Major Effects of the Disease?Early signs of the disease vary greatly from person to person. A common observation is that the earlier the symptoms appear, the faster the disease progresses. Family members may first notice that the individual experiences mood swings or becomes uncharacteristically irritable, apathetic, passive, depressed, or angry. These symptoms may lessen as the disease progresses or, in some individuals, may continue and include hostile outbursts or deep bouts of depression. HD may affect the individual's judgment, memory, and other cognitive functions. Early signs might include having trouble driving, learning new things, remembering a fact, answering a question, or making a decision. Some may even display changes in handwriting. As the disease progresses, concentration on intellectual tasks becomes increasingly difficult. In some individuals, the disease may begin with uncontrolled movements in the fingers, feet, face, or trunk. These movements—which are signs of chorea—often intensify when the person is anxious. HD can also begin with mild clumsiness or problems with balance. Some people develop choreic movements later, after the disease has progressed. They may stumble or appear uncoordinated. Chorea often creates serious problems with walking, increasing the likelihood of falls. The disease can reach the point where speech is slurred and vital functions, such as swallowing, eating, speaking, and especially walking, continue to decline. Some individuals cannot recognize other family members. Many, however, remain aware of their environment and are able to express emotions. Some physicians have employed a recently developed Unified HD Rating Scale, or UHDRS, to assess the clinical features, stages, and course of HD. In general, the duration of the illness ranges from 10 to 30 years. The most common causes of death are infection (most often pneumonia), injuries related to a fall, or other complications. At What Age Does HD Appear?The rate of disease progression and the age at onset vary from person to person. Adult-onset HD, with its disabling, uncontrolled movements, most often begins in middle age. There are, however, other variations of HD distinguished not just by age at onset but by a distinct array of symptoms. For example, some persons develop the disease as adults, but without chorea. They may appear rigid and move very little, or not at all, a condition called akinesia. Some individuals develop symptoms of HD when they are very young—before age 20. The terms "early-onset" or "juvenile" HD are often used to describe HD that appears in a young person. A common sign of HD in a younger individual is a rapid decline in school performance. Symptoms can also include subtle changes in handwriting and slight problems with movement, such as slowness, rigidity, tremor, and rapid muscular twitching, called myoclonus. Several of these symptoms are similar to those seen in Parkinson's disease, and they differ from the chorea seen in individuals who develop the disease as adults. These young individuals are said to have "akinetic-rigid" HD or the Westphal variant of HD. People with juvenile HD may also have seizures and mental disabilities. The earlier the onset, the faster the disease seems to progress. The disease progresses most rapidly in individuals with juvenile or early-onset HD, and death often follows within 10 years. Individuals with juvenile HD usually inherit the disease from their fathers. These individuals also tend to have the largest number of CAG repeats. The reason for this may be found in the process of sperm production. Unlike eggs, sperm are produced in the millions. Because DNA is copied millions of times during this process, there is an increased possibility for genetic mistakes to occur. To verify the link between the number of CAG repeats in the HD gene and the age at onset of symptoms, scientists studied a boy who developed HD symptoms at the age of two, one of the youngest and most severe cases ever recorded. They found that he had the largest number of CAG repeats of anyone studied so far—nearly 100. The boy's case was central to the identification of the HD gene and at the same time helped confirm that juveniles with HD have the longest segments of CAG repeats, the only proven correlation between repeat length and age at onset. A few individuals develop HD after age 55. Diagnosis in these people can be very difficult. The symptoms of HD may be masked by other health problems, or the person may not display the severity of symptoms seen in individuals with HD of earlier onset. These individuals may also show symptoms of depression rather than anger or irritability, or they may retain sharp control over their intellectual functions, such as memory, reasoning, and problem-solving. There is also a related disorder called senile chorea. Some elderly individuals display the symptoms of HD, especially choreic movements, but do not become demented, have a normal gene, and lack a family history of the disorder. Some scientists believe that a different gene mutation may account for this small number of cases, bu this has not been proven. How is HD Diagnosed?The great American folk singer and composer Woody Guthrie died on October 3, 1967, after suffering from HD for 13 years. He had been misdiagnosed, considered an alcoholic, and shuttled in and out of mental institutions and hospitals for years before being properly diagnosed. His case, sadly, is not extraordinary, although the diagnosis can be made easily by experienced neurologists. A neurologist will interview the individual intensively to obtain the medical history and rule out other conditions. A tool used by physicians to diagnose HD is to take the family history, sometimes called a pedigree or genealogy. It is extremely important for family members to be candid and truthful with a doctor who is taking a family history. The doctor will also ask about recent intellectual or emotional problems, which may be indications of HD, and will test the person's hearing, eye movements, strength, coordination, involuntary movements (chorea), sensation, reflexes, balance, movement, and mental status, and will probably order a number of laboratory tests as well. People with HD commonly have impairments in the way the eye follows or fixes on a moving target. Abnormalities of eye movements vary from person to person and differ, depending on the stage and duration of the illness. The discovery of the HD gene in 1993 resulted in a direct genetic test to make or confirm a diagnosis of HD in an individual who is exhibiting HD-like symptoms. Using a blood sample, the genetic test analyzes DNA for the HD mutation by counting the number of repeats in the HD gene region. Individuals who do not have HD usually have 28 or fewer CAG repeats. Individuals with HD usually have 40 or more repeats. A small percentage of individuals, however, have a number of repeats that fall within a borderline region (see table 1). Table 1
The physician may ask the individual to undergo a brain imaging test. Computed tomography (CT) and magnetic resonance imaging (MRI) provide excellent images of brain structures with little if any discomfort. Those with HD may show shrinkage of some parts of the brain—particularly two areas known as the caudate nuclei and putamen—and enlargement of fluid-filled cavities within the brain called ventricles. These changes do not definitely indicate HD, however, because they can also occur in other disorders. In addition, a person can have early symptoms of HD and still have a normal CT scan. When used in conjunction with a family history and record of clinical symptoms, however, CT can be an important diagnostic tool. Another technology for brain imaging includes positron emission tomography (PET,) which is important in HD research efforts but is not often needed for diagnosis. What is Presymptomatic Testing?Presymptomatic testing is used for people who have a family history of HD but have no symptoms themselves. If either parent had HD, the person's chance would be 50-50. In the past, no laboratory test could positively identify people carrying the HD gene—or those fated to develop HD—before the onset of symptoms. That situation changed in 1983, when a team of scientists supported by the NINDS located the first genetic marker for HD—the initial step in developing a laboratory test for the disease. A marker is a piece of DNA that lies near a gene and is usually inherited with it. Discovery of the first HD marker allowed scientists to locate the HD gene on chromosome 4. The marker discovery quickly led to the development of a presymptomatic test for some individuals, but this test required blood or tissue samples from both affected and unaffected family members in order to identify markers unique to that particular family. For this reason, adopted individuals, orphans, and people who had few living family members were unable to use the test. Discovery of the HD gene has led to a less expensive, scientifically simpler, and far more accurate presymptomatic test that is applicable to the majority of at-risk people. The new test uses CAG repeat length to detect the presence of the HD mutation in blood. This is discussed further in the next section. There are many complicating factors that reflect the complexity of diagnosing HD. In a small number of individuals with HD—1 to 3 percent—no family history of HD can be found. Some individuals may not be aware of their genetic legacy, or a family member may conceal a genetic disorder from fear of social stigma. A parent may not want to worry children, scare them, or deter them from marrying. In other cases, a family member may die of another cause before he or she begins to show signs of HD. Sometimes, the cause of death for a relative may not be known, or the family is not aware of a relative's death. Adopted children may not know their genetic heritage, or early symptoms in an individual may be too slight to attract attention. How is the Presymptomatic Test Conducted?An individual who wishes to be tested should contact the nearest testing center. (A list of such centers can be obtained from the Huntington Disease Society of America at 1-800-345-HDSA.) The testing process should include several components. Most testing programs include a neurological examination, pretest counseling, and follow up. The purpose of the neurological examination is to determine whether or not the person requesting testing is showing any clinical symptoms of HD. It is important to remember that if an individual is showing even slight symptoms of HD, he or she risks being diagnosed with the disease during the neurological examination, even before the genetic test. During pretest counseling, the individual will learn about HD, and about his or her own level of risk, about the testing procedure. The person will be told about the test's limitations, the accuracy of the test, and possible outcomes. He or she can then weigh the risks and benefits of testing and may even decide at that time against pursuing further testing. If a person decides to be tested, a team of highly trained specialists will be involved, which may include neurologists, genetic counselors, social workers, psychiatrists, and psychologists. This team of professionals helps the at-risk person decide if testing is the right thing to do and carefully prepares the person for a negative, positive, or inconclusive test result. Individuals who decide to continue the testing process should be accompanied to counseling sessions by a spouse, a friend, or a relative who is not at risk. Other interested family members may participate in the counseling sessions if the individual being tested so desires. The genetic testing itself involves donating a small sample of blood that is screened in the laboratory for the presence or absence of the HD mutation. Testing may require a sample of DNA from a closely related affected relative, preferably a parent, for the purpose of confirming the diagnosis of HD in the family. This is especially important if the family history for HD is unclear or unusual in some way. Results of the test should be given only in person and only to the individual being tested. Test results are confidential. Regardless of test results, follow up is recommended. In order to protect the interests of minors, including confidentiality, testing is not recommended for those under the age of 18 unless there is a compelling medical reason (for example, the child is exhibiting symptoms). Testing of a fetus (prenatal testing) presents special challenges and risks; in fact some centers do not perform genetic testing on fetuses. Because a positive test result using direct genetic testing means the at-risk parent is also a gene carrier, at-risk individuals who are considering a pregnancy are advised to seek genetic counseling prior to conception. Some at-risk parents may wish to know the risk to their fetus but not their own. In this situation, parents may opt for prenatal testing using linked DNA markers rather than direct gene testing. In this case, testing does not look for the HD gene itself but instead indicates whether or not the fetus has inherited a chromosome 4 from the affected grandparent or from the unaffected grandparent on the side of the family with HD. If the test shows that the fetus has inherited a chromosome 4 from the affected grandparent, the parents then learn that the fetus's risk is the same as the parent (50-50), but they learn nothing new about the parent's risk. If the test shows that the fetus has inherited a chromosome 4 from the unaffected grandparent, the risk to the fetus is very low (less than 1%) in most cases. Another option open to parents is in vitro fertilization with preimplantation screening. In this procedure, embryos are screened to determine which ones carry the HD mutation. Embryos determined not to have the HD gene mutation are then implanted in the woman's uterus. In terms of emotional and practical consequences, not only for the individual taking the test but for his or her entire family, testing is enormously complex and has been surrounded by considerable controversy. For example, people with a positive test result may risk losing health and life insurance, suffer loss of employment, and other liabilities. People undergoing testing may wish to cover the cost themselves, since coverage by an insurer may lead to loss of health insurance in the event of a positive result, although this may change in the future. With the participation of health professionals and people from families with HD, scientists have developed testing guidelines. All individuals seeking a genetic test should obtain a copy of these guidelines, either from their testing center or from the organizations listed on the card in the back of this brochure. These organizations have information on sites that perform testing using the established procedures and they strongly recommend that individuals avoid testing that does not adhere to these guidelines. How Does a Person Decide Whether to be Tested?The anxiety that comes from living with a 50 percent risk for HD can be overwhelming. How does a young person make important choices about long-term education, marriage, and children? How do older parents of adult children cope with their fears about children and grandchildren? How do people come to terms with the ambiguity and uncertainty of living at risk? Some individuals choose to undergo the test out of a desire for greater certainty about their genetic status. They believe the test will enable them to make more informed decisions about the future. Others choose not to take the test. They are able to make peace with the uncertainty of being at risk, preferring to forego the emotional consequences of a positive result, as well as possible losses of insurance and employment. There is no right or wrong decision, as each choice is highly individual. The guidelines for genetic testing for HD, discussed in the previous section, were developed to help people with this life-changing choice. Whatever the results of genetic testing, the at-risk individual and family members can expect powerful and complex emotional responses. The health and happiness of spouses, brothers and sisters, children, parents, and grandparents are affected by a positive test result, as are an individual's friends, work associates, neighbors, and others. Because receiving test results may prove to be devastating, testing guidelines call for continued counseling even after the test is complete and the results are known. Is There a Treatment for HD?Physicians may prescribe a number of medications to help control emotional and movement problems associated with HD. It is important to remember however, that while medicines may help keep these clinical symptoms under control, there is no treatment to stop or reverse the course of the disease. In August 2008 the U.S. Food and Drug Administration approved tetrabenazine to treat Huntington's chorea, making it the first drug approved for use in the United States to treat the disease. Antipsychotic drugs, such as haloperidol, or other drugs, such as clonazepam, may help to alleviate choreic movements and may also be used to help control hallucinations, delusions, and violent outbursts. Antipsychotic drugs, however, are not prescribed for another form of muscle contraction associated with HD, called dystonia, and may in fact worsen the condition, causing stiffness and rigidity. These medications may also have severe side effects, including sedation, and for that reason should be used in the lowest possible doses. For depression, physicians may prescribe fluoxetine, sertraline, nortriptyline, or other compounds. Tranquilizers can help control anxiety and lithium may be prescribed to combat pathological excitement and severe mood swings. Medications may also be needed to treat the severe obsessive-compulsive rituals of some individuals with HD. Most drugs used to treat the symptoms of HD have side effects such as fatigue, restlessness, or hyperexcitability. Sometimes it may be difficult to tell if a particular symptom, such as apathy or incontinence, is a sign of the disease or a reaction to medication. What Kind of Care Does the Individual with HD Need?Although a psychologist or psychiatrist, a genetic counselor, and other specialists may be needed at different stages of the illness, usually the first step in diagnosis and in finding treatment is to see a neurologist. While the family doctor may be able to diagnose HD, and may continue to monitor the individual's status, it is better to consult with a neurologist about management of the varied symptoms. Problems may arise when individuals try to express complex thoughts in words they can no longer pronounce intelligibly. It can be helpful to repeat words back to the person with HD so that he or she knows that some thoughts are understood. Sometimes people mistakenly assume that if individuals do not talk, they also do not understand. Never isolate individuals by not talking, and try to keep their environment as normal as possible. Speech therapy may improve the individual's ability to communicate. It is extremely important for the person with HD to maintain physical fitness as much as his or her condition and the course of the disease allows. Individuals who exercise and keep active tend to do better than those who do not. A daily regimen of exercise can help the person feel better physically and mentally. Although their coordination may be poor, individuals should continue walking, with assistance if necessary. Those who want to walk independently should be allowed to do so as long as possible, and careful attention should be given to keeping their environment free of hard, sharp objects. This will help ensure maximal independence while minimizing the risk of injury from a fall. Individuals can also wear special padding during walks to help protect against injury from falls. Some people have found that small weights around the ankles can help stability. Wearing sturdy shoes that fit well can help too, especially shoes without laces that can be slipped on or off easily. Impaired coordination may make it difficult for people with HD to feed themselves and to swallow. As the disease progresses, persons with HD may even choke. In helping individuals to eat, caregivers should allow plenty of time for meals. Food can be cut into small pieces, softened, or pureed to ease swallowing and prevent choking. While some foods may require the addition of thickeners, other foods may need to be thinned. Dairy products, in particular, tend to increase the secretion of mucus, which in turn increases the risk of choking. Some individuals may benefit from swallowing therapy, which is especially helpful if started before serious problems arise. Suction cups for plates, special tableware designed for people with disabilities, and plastic cups with tops can help prevent spilling. The individual's physician can offer additional advice about diet and about how to handle swallowing difficulties or gastrointestinal problems that might arise, such as incontinence or constipation. Caregivers should pay attention to proper nutrition so that the individual with HD takes in enough calories to maintain his or her body weight. Sometimes people with HD, who may burn as many as 5,000 calories a day without gaining weight, require five meals a day to take in the necessary number of calories. Physicians may recommend vitamins or other nutritional supplements. In a long-term care institution, staff will need to assist with meals in order to ensure that the individual's special caloric and nutritional requirements are met. Some individuals and their families choose to use a feeding tube; others choose not to. Individuals with HD are at special risk for dehydration and therefore require large quantities of fluids, especially during hot weather. Bendable straws can make drinking easier for the person. In some cases, water may have to be thickened with commercial additives to give it the consistency of syrup or honey. What Community Resources are Available?Individuals and families affected by HD can take steps to ensure that they receive the best advice and care possible. Physicians and state and local health service agencies can provide information on community resources and family support groups that may exist. Possible types of help include: Legal and social aid. HD affects a person's capacity to reason, make judgments, and handle responsibilities. Individuals may need help with legal affairs. Wills and other important documents should be drawn up early to avoid legal problems when the person with HD may no longer be able to represent his or her own interests. Family members should also seek out assistance if they face discrimination regarding insurance, employment, or other matters. Home care services. Caring for a person with HD at home can be exhausting, but part-time assistance with household chores or physical care of the individual can ease this burden. Domestic help, meal programs, nursing assistance, occupational therapy, or other home services may be available from federal, state, or local health service agencies. Recreation and work centers. Many people with HD are eager and able to participate in activities outside the home. Therapeutic work and recreation centers give individuals an opportunity to pursue hobbies and interests and to meet new people. Participation in these programs, including occupational, music, and recreational therapy, can reduce the person's dependence on family members and provides home caregivers with a temporary, much needed break. Group housing. A few communities have group housing facilities that are supervised by a resident attendant and that provide meals, housekeeping services, social activities, and local transportation services for residents. These living arrangements are particularly suited to the needs of individuals who are alone and who, although still independent and capable, risk injury when they undertake routine chores like cooking and cleaning. Institutional care. The individual's physical and emotional demands on the family may eventually become overwhelming. While many families may prefer to keep relatives with HD at home whenever possible, a long-term care facility may prove to be best. To hospitalize or place a family member in a care facility is a difficult decision; professional counseling can help families with this. Finding the proper facility can itself prove difficult. Organizations such as the Huntington's Disease Society of America (see listing on the Information Resources card in the back pocket of this brochure) may be able to refer the family to facilities that have met standards set for the care of individuals with HD. Very few of these exist however, and even fewer have experience with individuals with juvenile or early-onset HD who require special care because of their age and symptoms. What Research is Being Done?Although HD attracted considerable attention from scientists in the early 20th century, there was little sustained research on the disease until the late 1960s when the Committee to Combat Huntington's Disease and the Huntington's Chorea Foundation, later called the Hereditary Disease Foundation, first began to fund research and to campaign for federal funding. In 1977, Congress established the Commission for the Control of Huntington's Disease and Its Consequences, which made a series of important recommendations. Since then, Congress has provided consistent support for federal research, primarily through the National Institute of Neurological Disorders and Stroke, the government's lead agency for biomedical research on disorders of the brain and nervous system. The effort to combat HD proceeds along the following lines of inquiry, each providing important information about the disease: Basic neurobiology. Now that the HD gene has been located, investigators in the field of neurobiology-which encompasses the anatomy, physiology, and biochemistry of the nervous system-are continuing to study the HD gene with an eye toward understanding how it causes disease in the human body. Clinical research. Neurologists, psychologists, psychiatrists, and other investigators are improving our understanding of the symptoms and progression of the disease in patients while attempting to develop new therapeutics. Imaging. Scientific investigations using PET and other technologies are enabling scientists to see what the defective gene does to various structures in the brain and how it affects the body's chemistry and metabolism. Animal models. Laboratory animals, such as mice, are being bred in the hope of duplicating the clinical features of HD and can soon be expected to help scientists learn more about the symptoms and progression of the disease. Fetal tissue research. Investigators are implanting fetal tissue in rodents and nonhuman primates with the hope that success in this area will lead to understanding, restoring, or replacing functions typically lost by neuronal degeneration in individuals with HD. These areas of research are slowly converging and, in the process, are yielding important clues about the gene's relentless destruction of mind and body. The NINDS supports much of this exciting work. Molecular GeneticsFor 10 years, scientists focused on a segment of chromosome 4 and, in 1993, finally isolated the HD gene. The process of isolating the responsible gene—motivated by the desire to find a cure—was more difficult than anticipated. Scientists now believe that identifying the location of the HD gene is the first step on the road to a cure. Finding the HD gene involved an intense molecular genetics research effort with cooperating investigators from around the globe. In early 1993, the collaborating scientists announced they had isolated the unstable triplet repeat DNA sequence that has the HD gene. Investigators relied on the NINDS-supported Research Roster for Huntington's Disease, based at Indiana University in Indianapolis, to accomplish this work. First started in 1979, the roster contains data on many American families with HD, provides statistical and demographic data to scientists, and serves as a liaison between investigators and specific families. It provided the DNA from many families affected by HD to investigators involved in the search for the gene and was an important component in the identification of HD markers. For several years, NINDS-supported investigators involved in the search for the HD gene made yearly visits to the largest known kindred with HD—14,000 individuals—who live on Lake Maracaibo in Venezuela. The continuing trips enable scientists to study inheritance patterns of several interrelated families. The HD Gene and Its ProductAlthough scientists know that certain brain cells die in HD, the cause of their death is still unknown. Recessive diseases are usually thought to result from a gene that fails to produce adequate amounts of a substance essential to normal function. This is known as a loss-of-function gene. Some dominantly inherited disorders, such as HD, are thought to involve a gene that actively interferes with the normal function of the cell. This is known as a gain-of-function gene. How does the defective HD gene cause harm? The HD gene encodes a protein—which has been named huntingtin—the function of which is as yet unknown. The repeated CAG sequence in the gene causes an abnormal form of huntingtin to be made, in which the amino acid glutamine is repeated. It is the presence of this abnormal form, and not the absence of the normal form, that causes harm in HD. This explains why the disease is dominant and why two copies of the defective gene—one from both the mother and the father—do not cause a more serious case than inheritance from only one parent. With the HD gene isolated, NINDS-supported investigators are now turning their attention toward discovering the normal function of huntingtin and how the altered form causes harm. Scientists hope to reproduce, study, and correct these changes in animal models of the disease. Huntingtin is found everywhere in the body but only outside the cell's nucleus. Mice called "knockout mice" are bred in the laboratory to produce no huntingtin; they fail to develop past a very early embryo stage and quickly die. Huntingtin, scientists now know, is necessary for life. Investigators hope to learn why the abnormal version of the protein damages only certain parts of the brain. One theory is that cells in these parts of the brain may be supersensitive to this abnormal protein. Cell Death in HDAlthough the precise cause of cell death in HD is not yet known, scientists are paying close attention to the process of genetically programmed cell death that occurs deep within the brains of individuals with HD. This process involves a complex series of interlinked events leading to cellular suicide. Related areas of investigation include:
Several HD studies are aimed at understanding losses of nerve cells and receptors in HD. Neurons in the striatum are classified both by their size (large, medium, or small) and appearance (spiny or aspiny). Each type of neuron contains combinations of neurotransmitters. Scientists know that the destructive process of HD affects different subsets of neurons to varying degrees. The hallmark of HD, they are learning, is selective degeneration of medium-sized spiny neurons in the striatum. NINDS-supported studies also suggest that losses of certain types of neurons and receptors are responsible for different symptoms and stages of HD. What do these changes look like? In spiny neurons, investigators have observed two types of changes, each affecting the nerve cells' dendrites. Dendrites, found on every nerve cell, extend out from the cell body and are responsible for receiving messages from other nerve cells. In the intermediate stages of HD, dendrites grow out of control. New, incomplete branches form and other branches become contorted. In advanced, severe stages of HD, degenerative changes cause sections of dendrites to swell, break off, or disappear altogether. Investigators believe that these alterations may be an attempt by the cell to rebuild nerve cell contacts lost early in the disease. As the new dendrites establish connections, however, they may in fact contribute to nerve cell death. Such studies give compelling, visible evidence of the progressive nature of HD and suggest that new experimental therapies must consider the state of cellular degeneration. Scientists do not yet know exactly how these changes affect subsets of nerve cells outside the striatum. Animal Models of HDAs more is learned about cellular degeneration in HD, investigators hope to reproduce these changes in animal models and to find a way to correct or halt the process of nerve cell death. Such models serve the scientific community in general by providing a means to test the safety of new classes of drugs in nonhuman primates. NINDS-supported scientists are currently working to develop both nonhuman primate and mouse models to investigate nerve degeneration in HD and to study the effects of excitotoxicity on nerve cells in the brain. Investigators are working to build genetic models of HD using transgenic mice. To do this, scientists transfer the altered human HD gene into mouse embryos so that the animals will develop the anatomical and biological characteristics of HD. This genetic model of mouse HD will enable in-depth study of the disease and testing of new therapeutic compounds. Another idea is to insert into mice a section of DNA containing CAG repeats in the abnormal, disease gene range. This mouse equivalent of HD could allow scientists to explore the basis of CAG instability and its role in the disease process. Fetal Tissue ResearchA relatively new field in biomedical research involves the use of brain tissue grafts to study, and potentially treat, neurodegenerative disorders. In this technique, tissue that has degenerated is replaced with implants of fresh, fetal tissue, taken at the very early stages of development. Investigators are interested in applying brain tissue implants to HD research. Extensive animal studies will be required to learn if this technique could be of value in patients with HD. Clinical StudiesScientists are pursuing clinical studies that may one day lead to the development of new drugs or other treatments to halt the disease's progression. Examples of NINDS-supported investigations, using both asymptomatic and symptomatic individuals, include: Genetic studies on age of onset, inheritance patterns, and markers found within families. These studies may shed additional light on how HD is passed from generation to generation. Studies of cognition, intelligence, and movement. Studies of abnormal eye movements, both horizontal and vertical, and tests of patients' skills in a number of learning, memory, neuropsychological, and motor tasks may serve to identify when the various symptoms of HD appear and to characterize their range and severity. Clinical trials of drugs. Testing of various drugs may lead to new treatments and at the same time improve our understanding of the disease process in HD. Classes of drugs being tested include those that control symptoms, slow the rate of progression of HD, and block effects of excitotoxins, and those that might correct or replace other metabolic defects contributing to the development and progression of HD. ImagingNINDS-supported scientists are using positron emission tomography (PET) to learn how the gene affects the chemical systems of the body. PET visualizes metabolic or chemical abnormalities in the body, and investigators hope to ascertain if PET scans can reveal any abnormalities that signal HD. Investigators conducting HD research are also using PET to characterize neurons that have died and chemicals that are depleted in parts of the brain affected by HD. Like PET, a form of magnetic resonance imaging (MRI) called functional MRI can measure increases or decreases in certain brain chemicals thought to play a key role in HD. Functional MRI studies are also helping investigators understand how HD kills neurons in different regions of the brain. Imaging technologies allow investigators to view changes in the volume and structures of the brain and to pinpoint when these changes occur in HD. Scientists know that in brains affected by HD, the basal ganglia, cortex, and ventricles all show atrophy or other alterations. How Can I Help?In order to conduct HD research, investigators require samples of tissue or blood from families with HD. Access to individuals with HD and their families may be difficult however, because families with HD are often scattered across the country or around the world. A research project may need individuals of a particular age or gender or from a certain geographic area. Some scientists need only statistical data while others may require a sample of blood, urine, or skin from family members. All of these factors complicate the task of finding volunteers. The following NINDS-supported efforts bring together families with HD, voluntary health agencies, and scientists in an effort to advance science and speed a cure. The NINDS-sponsored HD Research Roster at the Indiana University Medical Center in Indianapolis, which was discussed earlier, makes research possible by matching scientists with patient and family volunteers. The first DNA bank was established through the roster. Although the gene has already been located, DNA from individuals who have HD is still of great interest to investigators. Of continuing interest are twins, unaffected individuals who have affected offspring, and individuals with two defective HD genes, one from each parent-a very rare occurrence. Participation in the roster and in specific research projects is voluntary and confidential. For more information about the roster and DNA bank, contact: Indiana University Medical Center The NINDS supports two national brain specimen banks. These banks supply research scientists around the world with nervous system tissue from patients with neurological and psychiatric disorders. They need tissue from patients with HD so that scientists can study and understand the disorder. Those who may be interested in donating should write to: Human Brain and Spinal Fluid Resource Center Francine M. Benes, M.D., Ph.D., Director What is the Role of Voluntary Organizations?Private organizations have been a mainstay of support and guidance for at-risk individuals, people with HD, and their families. These organizations vary in size and emphasis, but all are concerned with helping individuals and their families, educating lay and professional audiences about HD, and promoting medical research on the disorder. Some voluntary health agencies support scientific workshops and research and some have newsletters and local chapters throughout the country. These agencies enable families, health professionals, and investigators to exchange information, learn of available services and benefits, and work toward common goals. The organizations listed on the Information Resources card in the back pocket of this brochure welcome inquiries from the public. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
akinesia-decreased body movements. NIH Publication No. 98-49
|
|
Introduction
Why Does it Hurt? When Should You See a Physician? What Tests Are Used to Diagnose Headache? What Are Migraine Headaches? How is Migraine Headache Treated? Besides Migraine, What Are Other Types of Vascular Headaches? What Are Muscle-Contraction Headaches? When is Headache a Warning of a More Serious Condition? What Causes Headache in Children? Conclusion Where can I get more information? Glossary IntroductionFor 2 years, Jim suffered the excruciating pain of cluster headaches. Night after night he paced the floor, the pain driving him to constant motion. He was only 48 years old when the clusters forced him to quit his job as a systems analyst. One year later, his headaches are controlled. The credit for Jim's recovery belongs to the medical staff of a headache clinic. Physicians there applied the latest research findings on headache, and prescribed for Jim a combination of new drugs. Joan was a victim of frequent migraine. Her headaches lasted 2 days. Nauseous and weak, she stayed in the dark until each attack was over. Today, although migraine still interferes with her life, she has fewer attacks and less severe headaches than before. A specialist prescribed an antimigraine program for Joan that included improved drug therapy, a new diet and relaxation training. An avid reader, Peggy couldn't put down the new mystery thriller. After 4 hours of reading slumped in bed, she knew she had overdone it. Her tensed head and neck muscles felt as if they were being squeezed between two giant hands. But for Peggy, the muscle-contraction headache and neck pain were soon relieved by a hot shower and aspirin. Understanding why headaches occur and improving headache treatment
are among the research goals of the National Institute of Neurological
Disorders and Stroke (NINDS). As the leading supporter of brain
research in the Federal Government, the NINDS also supports and
conducts studies to improve the diagnosis of headaches and to
find ways to prevent them. Why Does it Hurt?What hurts when you have a headache? The bones of the skull and tissues of the brain itself never hurt, because they lack pain-sensitive nerve fibers. Several areas of the head can hurt, including a network of nerves which extends over the scalp and certain nerves in the face, mouth, and throat. Also sensitive to pain, because they contain delicate nerve fibers, are the muscles of the head and blood vessels found along the surface and at the base of the brain. The ends of these pain-sensitive nerves, called nociceptors, can be stimulated by stress, muscular tension, dilated blood vessels, and other triggers of headache. Once stimulated, a nociceptor sends a message up the length of the nerve fiber to the nerve cells in the brain, signaling that a part of the body hurts. The message is determined by the location of the nociceptor. A person who suddenly realizes "My toe hurts," is responding to nociceptors in the foot that have been stimulated by the stubbing of a toe. A number of chemicals help transmit pain-related information to the brain. Some of these chemicals are natural painkilling proteins called endorphins, Greek for "the morphine within." One theory suggests that people who suffer from severe headache and other types of chronic pain have lower levels of endorphins than people who are generally pain free. When Should You See a Physician?Not all headaches require medical attention. Some result from missed meals or occasional muscle tension and are easily remedied. But some types of headache are signals of more serious disorders, and call for prompt medical care. These include:
A headache sufferer usually seeks help from a family practitioner. If the problem is not relieved by standard treatments, the patient may then be referred to a specialist - perhaps an internist or neurologist. Additional referrals may be made to psychologists. What Tests Are Used to Diagnose Headache?Diagnosing a headache is like playing Twenty Questions. Experts agree that a detailed question-and-answer session with a patient can often produce enough information for a diagnosis. Many types of headaches have clear-cut symptoms which fall into an easily recognizable pattern. Patients may be asked: How often do you have headaches? Where is the pain? How long do the headaches last? When did you first develop headaches? The patient's sleep habits and family and work situations may also be probed. Most physicians will also obtain a full medical history from the patient, inquiring about past head trauma or surgery, eye strain, sinus problems, dental problems, difficulties with opening and closing of the jaw, and the use of medications. This may be enough to suggest strongly that the patient has migraine or cluster headaches. A complete and careful physical and neurological examination will exclude many possibilities and the suspicion of aneurysm, meningitis, or certain brain tumors. A blood test may be ordered to screen for thyroid disease, anemia, or infections which might cause a headache. A test called an electroencephalogram (EEG) may be given to measure brain activity. EEG's can indicate a malfunction in the brain, but they cannot usually pinpoint a problem that might be causing a headache. A physician may suggest that a patient with unusual headaches undergo a computed tomographic (CT) scan and/or a magnetic resonance imaging (MRI) scan. The scans enable the physician to distinguish, for example, between a bleeding blood vessel in the brain and a brain tumor, and are important diagnostic tools in cases of headache associated with brain lesions or other serious disease. CT scans produce X-ray images of the brain that show structures or variations in the density of different types of tissue. MRI scans use magnetic fields and radio waves to produce an image that provides information about the structure and biochemistry of the brain. If an aneurysm-an abnormal ballooning of a blood vessel-is suspected, a physician may order a CT scan to examine for blood and then an angiogram. In this test, a special fluid which can be seen on an X-ray is injected into the patient and carried in the bloodstream to the brain to reveal any abnormalities in the blood vessels there. A physician analyzes the results of all these diagnostic tests along with a patient's medical history and examination in order to arrive at a diagnosis. Headaches are diagnosed as
Vascular headaches - a group that includes the well-known migraine - are so named because they are thought to involve abnormal function of the brain's blood vessels or vascular system. Muscle contraction headaches appear to involve the tightening or tensing of facial and neck muscles. Traction and inflammatory headaches are symptoms of other disorders, ranging from stroke to sinus infection. Some people have more than one type of headache. What Are Migraine Headaches?The most common type of vascular headache is migraine. Migraine headaches are usually characterized by severe pain on one or both sides of the head, an upset stomach, and at times disturbed vision. Former basketball star Kareem Abdul-Jabbar remembers experiencing his first migraine at age 14. The pain was unlike the discomfort of his previous mild headaches. "When I got this one I thought, 'This is a headache'," he says. "The pain was intense and I felt nausea and a great sensitivity to light. All I could think about was when it would stop. I sat in a dark room for an hour and it passed." Symptoms of migraine. Abdul-Jabbar's sensitivity to light is a standard symptom of the two most prevalent types of migraine-caused headache: classic and common. The major difference between the two types is the appearance of neurological symptoms 10 to 30 minutes before a classic migraine attack. These symptoms are called an aura. The person may see flashing lights or zigzag lines, or may temporarily lose vision. Other classic symptoms include speech difficulty, weakness of an arm or leg, tingling of the face or hands, and confusion. The pain of a classic migraine headache may be described as intense, throbbing, or pounding and is felt in the forehead, temple, ear, jaw, or around the eye. Classic migraine starts on one side of the head but may eventually spread to the other side. An attack lasts 1 to 2 pain-wracked days. Common migraine - a term that reflects the disorder's greater occurrence in the general population - is not preceded by an aura. But some people experience a variety of vague symptoms beforehand, including mental fuzziness, mood changes, fatigue, and unusual retention of fluids. During the headache phase of a common migraine, a person may have diarrhea and increased urination, as well as nausea and vomiting. Common migraine pain can last 3 or 4 days. Both classic and common migraine can strike as often as several times a week, or as rarely as once every few years. Both types can occur at any time. Some people, however, experience migraines at predictable times - for example, near the days of menstruation or every Saturday morning after a stressful week of work. The migraine process. Research scientists are unclear about the precise cause of migraine headaches. There seems to be general agreement, however, that a key element is blood flow changes in the brain. People who get migraine headaches appear to have blood vessels that overreact to various triggers. Scientists have devised one theory of migraine which explains these blood flow changes and also certain biochemical changes that may be involved in the headache process. According to this theory, the nervous system responds to a trigger such as stress by causing a spasm of the nerve-rich arteries at the base of the brain. The spasm closes down or constricts several arteries supplying blood to the brain, including the scalp artery and the carotid or neck arteries. As these arteries constrict, the flow of blood to the brain is reduced. At the same time, blood-clotting particles called platelets clump together-a process which is believed to release a chemical called serotonin. Serotonin acts as a powerful constrictor of arteries, further reducing the blood supply to the brain. Reduced blood flow decreases the brain's supply of oxygen. Symptoms signaling a headache, such as distorted vision or speech, may then result, similar to symptoms of stroke. Reacting to the reduced oxygen supply, certain arteries within the brain open wider to meet the brain's energy needs. This widening or dilation spreads, finally affecting the neck and scalp arteries. The dilation of these arteries triggers the release of pain-producing substances called prostaglandins from various tissues and blood cells. Chemicals which cause inflammation and swelling, and substances which increase sensitivity to pain, are also released. The circulation of these chemicals and the dilation of the scalp arteries stimulate the pain-sensitive nociceptors. The result, according to this theory: a throbbing pain in the head. Women and migraine. Although both males and females seem to be equally affected by migraine, the condition is more common in adult women. Both sexes may develop migraine in infancy, but most often the disorder begins between the ages of 5 and 35. The relationship between female hormones and migraine is still unclear. Women may have "menstrual migraine" - headaches around the time of their menstrual period - which may disappear during pregnancy. Other women develop migraine for the first time when they are pregnant. Some are first affected after menopause. The effect of oral contraceptives on headaches is perplexing. Scientists report that some women with migraine who take birth control pills experience more frequent and severe attacks. However, a small percentage of women have fewer and less severe migraine headaches when they take birth control pills. And normal women who do not suffer from headaches may develop migraines as a side effect when they use oral contraceptives. Investigators around the world are studying hormonal changes in women with migraine in the hope of identifying the specific ways these naturally occurring chemicals cause headaches. Triggers of headache. Although many sufferers have a family history of migraine, the exact hereditary nature of this condition is still unknown. People who get migraines are thought to have an inherited abnormality in the regulation of blood vessels. "It's like a cocked gun with a hair trigger," explains one specialist. "A person is born with a potential for migraine and the headache is triggered by things that are really not so terrible." These triggers include stress and other normal emotions, as well as biological and environmental conditions. Fatigue, glaring or flickering lights, changes in the weather, and certain foods can set off migraine. It may seem hard to believe that eating such seemingly harmless foods as yogurt, nuts, and lima beans can result in a painful migraine headache. However, some scientists believe that these foods and several others contain chemical substances, such as tyramine, which constrict arteries - the first step of the migraine process. Other scientists believe that foods cause headaches by setting off an allergic reaction in susceptible people. While a food-triggered migraine usually occurs soon after eating, other triggers may not cause immediate pain. Scientists report that people can develop migraine not only during a period of stress but also afterwards when their vascular systems are still reacting. For example, migraines that wake people up in the middle of the night are believed to result from a delayed reaction to stress. Other forms of migraine. In addition to classic and common, migraine headache can take several other forms: Patients with hemiplegic migraine have temporary paralysis on one side of the body, a condition known as hemiplegia. Some people may experience vision problems and vertigo - a feeling that the world is spinning. These symptoms begin 10 to 90 minutes before the onset of headache pain. In ophthalmoplegic migraine, the pain is around the eye and is associated with a droopy eyelid, double vision, and other problems with vision. Basilar artery migraine involves a disturbance of a major brain artery at the base of the brain. Preheadache symptoms include vertigo, double vision, and poor muscular coordination. This type of migraine occurs primarily in adolescent and young adult women and is often associated with the menstrual cycle. Benign exertional headache is brought on by running, lifting, coughing, sneezing, or bending. The headache begins at the onset of activity, and pain rarely lasts more than several minutes. Status migrainosus is a rare and severe type of migraine that can last 72 hours or longer. The pain and nausea are so intense that people who have this type of headache must be hospitalized. The use of certain drugs can trigger status migrainosus. Neurologists report that many of their status migrainosus patients were depressed and anxious before they experienced headache attacks. Headache-free migraine is characterized by such migraine symptoms as visual problems, nausea, vomiting, constipation, or diarrhea. Patients, however, do not experience head pain. Headache specialists have suggested that unexplained pain in a particular part of the body, fever, and dizziness could also be possible types of headache-free migraine. How is Migraine Headache Treated?During the Stone Age, pieces of a headache sufferer's skull were cut away with flint instruments to relieve pain. Another unpleasant remedy used in the British Isles around the ninth Century involved drinking "the juice of elderseed, cow's brain, and goat's dung dissolved in vinegar." Fortunately, today's headache patients are spared such drastic measures. Drug therapy, biofeedback training, stress reduction, and elimination of certain foods from the diet are the most common methods of preventing and controlling migraine and other vascular headaches. Joan, the migraine sufferer, was helped by treatment with a combination of an antimigraine drug and diet control. Regular exercise, such as swimming or vigorous walking, can also reduce the frequency and severity of migraine headaches. Joan found that whirlpool and yoga baths helped her relax. During a migraine headache, temporary relief can sometimes be obtained by applying cold packs to the head or by pressing on the bulging artery found in front of the ear on the painful side of the head. Drug therapy. There are two ways to approach the treatment of migraine headache with drugs: prevent the attacks, or relieve symptoms after the headache occurs. For infrequent migraine, drugs can be taken at the first sign of a headache in order to stop it or to at least ease the pain. People who get occasional mild migraine may benefit by taking aspirin or acetaminophen at the start of an attack. Aspirin raises a person's tolerance to pain and also discourages clumping of blood platelets. Small amounts of caffeine may be useful if taken in the early stages of migraine. But for most migraine sufferers who get moderate to severe headaches, and for all cluster headache patients (see section "Besides Migraine, What Are Other Types of Vascular Headaches?"), stronger drugs may be necessary to control the pain. Several drugs for the prevention of migraine have been developed in recent years, including serotonin agonists which mimic the action of this key brain chemical. One of the most commonly used drugs for the relief of classic and common migraine symptoms is sumatriptan, which binds to serotonin receptors. For optimal benefit, the drug is taken during the early stages of an attack. If a migraine has been in progress for about an hour after the drug is taken, a repeat dose can be given. Physicians caution that sumatriptan should not be taken by people who have angina pectoris, basilar migraine, severe hypertension, or vascular, or liver disease. Another migraine drug is ergotamine tartrate, a vasoconstrictor which helps counteract the painful dilation stage of the headache. Other drugs that constrict dilated blood vessels or help reduce blood vessel inflammation also are available. For headaches that occur three or more times a month, preventive treatment is usually recommended. Drugs used to prevent classic and common migraine include methysergide maleate, which counteracts blood vessel constriction; propranolol hydrochloride, which stops blood vessel dilation; amitriptyline, an antidepressant; valproic acid, an anticonvulsant; and verapamil, a calcium channel blocker. Antidepressants called MAO inhibitors also prevent migraine. These drugs block an enzyme called monoamine oxidase which normally helps nerve cells absorb the artery-constricting brain chemical, serotonin. MAO inhibitors can have potentially serious side effects - particularly if taken while ingesting foods or beverages that contain tyramine, a substance that constricts arteries. Many antimigraine drugs can have adverse side effects. But like most medicines they are relatively safe when used carefully and under a physician's supervision. To avoid long-term side effects of preventive medications, headache specialists advise patients to reduce the dosage of these drugs and then stop taking them as soon as possible. Biofeedback and relaxation training. Drug therapy for migraine is often combined with biofeedback and relaxation training. Biofeedback refers to a technique that can give people better control over such body function indicators as blood pressure, heart rate, temperature, muscle tension, and brain waves. Thermal biofeedback allows a patient to consciously raise hand temperature. Some patients who are able to increase hand temperature can reduce the number and intensity of migraines. The mechanisms underlying these self-regulation treatments are being studied by research scientists. "To succeed in biofeedback," says a headache specialist, "you must be able to concentrate and you must be motivated to get well." A patient learning thermal biofeedback wears a device which transmits the temperature of an index finger or hand to a monitor. While the patient tries to warm his hands, the monitor provides feedback either on a gauge that shows the temperature reading or by emitting a sound or beep that increases in intensity as the temperature increases. The patient is not told how to raise hand temperature, but is given suggestions such as "Imagine your hands feel very warm and heavy." "I have a good imagination," says one headache sufferer who traded in her medication for thermal biofeedback. The technique decreased the number and severity of headaches she experienced. In another type of biofeedback called electromyographic or EMG training, the patient learns to control muscle tension in the face, neck, and shoulders. Either kind of biofeedback may be combined with relaxation training, during which patients learn to relax the mind and body. Biofeedback can be practiced at home with a portable monitor. But the ultimate goal of treatment is to wean the patient from the machine. The patient can then use biofeedback anywhere at the first sign of a headache. The antimigraine diet. Scientists estimate that a small percentage of migraine sufferers will benefit from a treatment program focused solely on eliminating headache-provoking foods and beverages. Other migraine patients may be helped by a diet to prevent low blood sugar. Low blood sugar, or hypoglycemia, can cause headache. This condition can occur after a period without food: overnight, for example, or when a meal is skipped. People who wake up in the morning with a headache may be reacting to the low blood sugar caused by the lack of food overnight. Treatment for headaches caused by low blood sugar consists of scheduling smaller, more frequent meals for the patient. A special diet designed to stabilize the body's sugar-regulating system is sometimes recommended. For the same reason, many specialists also recommend that migraine patients avoid oversleeping on weekends. Sleeping late can change the body's normal blood sugar level and lead to a headache. Besides Migraine, What Are Other Types of Vascular Headaches?After migraine, the most common type of vascular headache is the toxic headache produced by fever. Pneumonia, measles, mumps, and tonsillitis are among the diseases that can cause severe toxic vascular headaches. Toxic headaches can also result from the presence of foreign chemicals in the body. Other kinds of vascular headaches include "clusters," which cause repeated episodes of intense pain, and headaches resulting from a rise in blood pressure. Chemical culprits. Repeated exposure to nitrite compounds can result in a dull, pounding headache that may be accompanied by a flushed face. Nitrite, which dilates blood vessels, is found in such products as heart medicine and dynamite, but is also used as a chemical to preserve meat. Hot dogs and other processed meats containing sodium nitrite can cause headaches. Eating foods prepared with monosodium glutamate (MSG) can result in headache. Soy sauce, meat tenderizer, and a variety of packaged foods contain this chemical which is touted as a flavor enhancer. Headache can also result from exposure to poisons, even common household varieties like insecticides, carbon tetrachloride, and lead. Children who ingest flakes of lead paint may develop headaches. So may anyone who has contact with lead batteries or lead-glazed pottery. Artists and industrial workers may experience headaches after exposure to materials that contain chemical solvents. These solvents, like benzene, are found in turpentine, spray adhesives, rubber cement, and inks. Drugs such as amphetamines can cause headaches as a side effect. Another type of drug-related headache occurs during withdrawal from long-term therapy with the antimigraine drug ergotamine tartrate. Jokes are often made about alcohol hangovers but the headache associated with "the morning after" is no laughing matter. Fortunately, there are several suggested treatments for the pain. The hangover headache may also be reduced by taking honey, which speeds alcohol metabolism, or caffeine, a constrictor of dilated arteries. Caffeine, however, can cause headaches as well as cure them. Heavy coffee drinkers often get headaches when they try to break the caffeine habit. Cluster headaches. Cluster headaches are a rare form of headache notable for their extreme pain and their pattern of occurring in "clusters", usually at the same time(s) of the day for several weeks. A cluster headache usually begins suddenly with excruciating pain on one side of the head, often behind or around one eye. In some individuals, it may be preceded by a migraine-like "aura." The pain usually peaks over the next 5 to 10 minutes, and then continues at that intensity for up to an hour or two before going away. People with cluster headaches describe the pain as piercing and unbearable. The nose and the eye on the affected side of the face may also get red, swollen, and runny, and some people will experience nausea, restlessness and agitation, or sensitivities to light, sound, or smell. Most affected individuals have one to three cluster headaches a day and two cluster periods a year, separated by periods of freedom from symptoms. A small group of people develop a chronic form of the disorder, characterized by bouts of cluster headaches that can go on for years with only brief periods (2 weeks or less) of remission. Cluster headaches generally begin between the ages of 20 and 50, although the syndrome can also start in childhood or late in life. Males are much more likely than females to develop cluster headaches. Alcohol (especially red wine) provokes attacks in more than half of those with cluster headaches, but has no effect once the cluster period ends. Cluster headaches are also strongly associated with cigarette smoking. Scientists aren't sure what causes the disorder. The tendency of cluster headaches to occur during the same time(s) from day to day, and more often at night than during the daylight hours, suggests they could be caused by irregularities in the body’s circadian rhythms, which are controlled by the brain and a family of hormones that regulate the sleep-wake cycle. There are medications available to lessen the pain of a cluster headache and suppress future attacks. Oxygen inhalation and triptan drugs (such as those used to treat migraine) administered as a tablet, nasal spray, or injection can provide quick relief from acute cluster headache pain. Lidocaine nasal spray, which numbs the nose and nostrils, may also be effective. Ergotamine and corticosteroids such as prednisone and dexamethasone may be prescribed to break the cluster cycle and then tapered off once headaches end. Verapamil may be used preventively to decrease the frequency and pain level of attacks. Lithium, valproic acid, and topiramate are sometimes also used preventively. Painful pressure . Chronic high blood pressure can cause headache, as can rapid rises in blood pressure like those experienced during anger, vigorous exercise, or sexual excitement. The
severe "orgasmic headache" occurs right before orgasm and
is believed to be a vascular headache. Since sudden rupture of
a cerebral blood vessel can occur, this type of headache should
be evaluated by a doctor. What Are Muscle-Contraction Headaches?It's 5:00 p.m. and your boss has just asked you to prepare a 20-page briefing paper. Due date: tomorrow. You're angry and tired and the more you think about the assignment, the tenser you become. Your teeth clench, your brow wrinkles, and soon you have a splitting tension headache. Tension headache is named not only for the role of stress in triggering the pain, but also for the contraction of neck, face, and scalp muscles brought on by stressful events. Tension headache is a severe but temporary form of muscle-contraction headache. The pain is mild to moderate and feels like pressure is being applied to the head or neck. The headache usually disappears after the period of stress is over. Ninety percent of all headaches are classified as tension/muscle contraction headaches. By contrast, chronic muscle-contraction headaches can last for weeks, months, and sometimes years. The pain of these headaches is often described as a tight band around the head or a feeling that the head and neck are in a cast. "It feels like somebody is tightening a giant vise around my head," says one patient. The pain is steady, and is usually felt on both sides of the head. Chronic muscle-contraction headaches can cause sore scalps - even combing one's hair can be painful. In the past, many scientists believed that the primary cause of the pain of muscle-contraction headache was sustained muscle tension. However, a growing number of authorities now believe that a far more complex mechanism is responsible. Occasionally, muscle-contraction headaches will be accompanied by nausea, vomiting, and blurred vision, but there is no preheadache syndrome as with migraine. Muscle-contraction headaches have not been linked to hormones or foods, as has migraine, nor is there a strong hereditary connection. Research has shown that for many people, chronic muscle-contraction headaches are caused by depression and anxiety. These people tend to get their headaches in the early morning or evening when conflicts in the office or home are anticipated. Emotional factors are not the only triggers of muscle-contraction headaches. Certain physical postures that tense head and neck muscles - such as holding one's chin down while reading - can lead to head and neck pain. So can prolonged writing under poor light, or holding a phone between the shoulder and ear, or even gum-chewing. More serious problems that can cause muscle-contraction headaches include degenerative arthritis of the neck and temporomandibular joint dysfunction, or TMD. TMD is a disorder of the joint between the temporal bone (above the ear) and the mandible or lower jaw bone. The disorder results from poor bite and jaw clenching. Treatment for muscle-contraction headache varies. The first consideration is to treat any specific disorder or disease that may be causing the headache. For example, arthritis of the neck is treated with anti-inflammatory medication and TMD may be helped by corrective devices for the mouth and jaw. Acute tension headaches not associated with a disease are treated with analgesics like aspirin and acetaminophen. Stronger analgesics, such as propoxyphene and codeine, are sometimes prescribed. As prolonged use of these drugs can lead to dependence, patients taking them should have periodic medical checkups and follow their physicians' instructions carefully. Nondrug therapy for chronic muscle-contraction headaches includes biofeedback, relaxation training, and counseling. A technique called cognitive restructuring teaches people to change their attitudes and responses to stress. Patients might be encouraged, for example, to imagine that they are coping successfully with a stressful situation. In progressive relaxation therapy, patients are taught to first tense and then relax individual muscle groups. Finally, the patient tries to relax his or her whole body. Many people imagine a peaceful scene - such as lying on the beach or by a beautiful lake. Passive relaxation does not involve tensing of muscles. Instead, patients are encouraged to focus on different muscles, suggesting that they relax. Some people might think to themselves, Relax or My muscles feel warm. People with chronic muscle-contraction headaches my also be helped by taking antidepressants or MAO inhibitors. Mixed muscle-contraction and migraine headaches are sometimes treated with barbiturate compounds, which slow down nerve function in the brain and spinal cord. People who suffer infrequent muscle-contraction headaches may benefit from a hot shower or moist heat applied to the back of the neck. Cervical collars are sometimes recommended as an aid to good posture. Physical therapy, massage, and gentle exercise of the neck may also be helpful. When is Headache a Warning of a More Serious Condition?Like other types of pain, headaches can serve as warning signals of more serious disorders. This is particularly true for headaches caused by traction or inflammation. Traction headaches can occur if the pain-sensitive parts of the head are pulled, stretched, or displaced, as, for example, when eye muscles are tensed to compensate for eyestrain. Headaches caused by inflammation include those related to meningitis as well as those resulting from diseases of the sinuses, spine, neck, ears, and teeth. Ear and tooth infections and glaucoma can cause headaches. In oral and dental disorders, headache is experienced as pain in the entire head, including the face. These headaches are treated by curing the underlying problem. This may involve surgery, antibiotics, or other drugs. Characteristics of the various types of more serious traction and inflammatory headaches vary by disorder:
What Causes Headache in Children?Like adults, children experience the infections, trauma, and stresses that can lead to headaches. In fact, research shows that as young people enter adolescence and encounter the stresses of puberty and secondary school, the frequency of headache increases. Migraine headaches often begin in childhood or adolescence. According to recent surveys, as many as half of all schoolchildren experience some type of headache. Children with migraine often have nausea and excessive vomiting. Some children have periodic vomiting, but no headache - the so-called abdominal migraine. Research scientists have found that these children usually develop headaches when they are older. Physicians have many drugs to treat migraine in children. Different classes that may be tried include analgesics, antiemetics, anticonvulsants, beta-blockers, and sedatives. A diet may also be prescribed to protect the child from foods that trigger headache. Sometimes psychological counseling or even psychiatric treatment for the child and the parents is recommended Childhood headache can be a sign of depression. Parents should alert the family pediatrician if a child develops headaches along with other symptoms such as a change in mood or sleep habits. Antidepressant medication and psychotherapy are effective treatments for childhood depression and related headache. ConclusionIf you suffer from headaches and none of the standard treatments help, do not despair. Some people find that their headaches disappear once they deal with a troubled marriage, pass their certifying board exams, or resolve some other stressful problem. Others find that if they control their psychological reaction to stress, the headaches disappear. "I had migraines for several years," says one woman, "and then they went away. I think it was because I lowered my personal goals in life. Today, even though I have 100 things to do at night, I don't worry about it. I learned to say no." For those who cannot say no, or who get headaches anyway, today's
headache research offers hope. The work of NINDS-supported scientists
around the world promises to improve our understanding of this
complex disorder and provide better tools to treat it. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
angiography-an imaging technique that provides a picture, called an angiogram, of blood vessels. aura-a symptom of classic migraine headache in which the patient sees flashing lights or zigzag lines, or may temporarily lose vision basilar artery migraine-migraine, occurring primarily in young women and often associated with the menstrual cycle, that involves a disturbance of a major brain artery. Symptoms include vertigo, double vision, and poor muscular coordination. benign exertional headache-headache brought on by running, lifting, coughing, sneezing, or bending. biofeedback-a technique in which patients are trained to gain some voluntary control over certain physiological conditions, such as blood pressure and muscle tension, to promote relaxation. Thermal biofeedback helps patients consciously raise hand temperature, which can sometimes reduce the number and intensity of migraines. cluster headaches-intensely painful headaches-occurring suddenly and lasting between 30 and 45 minutes-named for their repeated occurrence in groups or clusters. They begin as minor pain around one eye and eventually spread to that side of the face. computer tomography (CT)-an imaging technique that uses X-rays and computer analysis to provide a picture of body tissues and structures. dihydroergotamine-a drug that is given by injection to treat cluster headaches. It is a form of the antimigraine drug ergotamine tartrate. electroencephalogram (EEG)-a technique for recording electrical activity in the brain. electromyography (EMG)-a special recording technique that detects electric activity in muscle. Patients are sometimes offered a type of biofeedback called EMG training, in which they learn to control muscle tension in the face, neck, and shoulders. endorphins-naturally occurring painkilling chemicals. Some scientists theorize that people who suffer from severe headache have lower levels of endorphins than people who are generally pain free. ergotamine tartrate-a drug that is used to control the painful dilation stage of migraine. hemiplegic migraine-a type of migraine causing temporary paralysis on one side of the body (hemiplegia) inflammatory headache-a headache that is a symptom of another disorder, such as sinus infection, and is treated by curing the underlying problem. magnetic resonance imaging (MRI)-an imaging technique that uses radio waves, magnetic fields, and computer analysis to provide a picture of body tissues and structures. migraine-a vascular headache believed to be caused by blood flow changes and certain chemical changes in the brain leading to a cascade of events - including constriction of arteries supplying blood to the brain and the release of certain brain chemicals - that result in severe head pain, stomach upset, and visual disturbances. muscle-contraction headaches-headaches caused primarily by sustained muscle tension or, possibly, by restricted blood flow to the brain. Two forms of muscle-contraction headache are tension headache, induced by stress, and chronic muscle-contraction headache, which can last for extended periods, involves steady pain, and is usually felt on both sides of the head. nociceptors-the endings of pain-sensitive nerves that, when stimulated by stress, muscular tension, dilated blood vessels, or other triggers, send messages up the nerve fibers to nerve cells in the brain, signaling that a part of the body hurts. ophthalmoplegic migraine-a form of migraine felt around the eye and associated with a droopy eyelid, double vision, and other sight problems. prostaglandins-naturally occurring pain-producing substances thought to be implicated in migraine attacks. Their release is triggered by the dilation of arteries. Prostaglandins are extremely potent chemicals involved in a diverse group of physiological processes. serotonin-a key neurotransmitter that acts as a powerful constrictor of arteries, reducing the blood supply to the brain and contributing to the pain of headache. sinusitis-an infection, either viral or bacterial, of the sinus cavities. The infection leads to inflammation of these cavities, causing pain and sometimes headache. sumatriptan-a commonly used migraine drug that binds to receptors for the neurotransmitter serotonin. status migrainosus-a rare, sustained, and severe type of migraine, characterized by intense pain and nausea and often leading to hospitalization of the patient. thermography-a technique sometimes used for diagnosing headache in which an infrared camera converts skin temperature into a color picture, called a thermogram, with different degrees of heat appearing as different colors. temporomandibular joint dysfunction-a disorder of the joint between the temporal bone (above the ear) and the lower jaw bone that can cause muscle-contraction headaches. tic douloureux-see trigeminal neuralgia traction headaches-headaches caused by pulling or stretching pain-sensitive parts of the head, as, for example, when eye muscles are tensed to compensate for eyestrain. trigeminal neuralgia-a condition resulting from a disorder of the trigeminal nerve. Symptoms are headache and intense facial pain that comes in short, excruciating jabs. vascular headaches- headaches caused by abnormal
function of the brain's blood vessels or vascular system. Migraine
is a type of vascular headache. NIH Publication No. 02-158
|
|
Heads
Up Download
the pdf file: Download
the pdf file: |
|
Acute
Management of Mild Traumatic Brain Injury in Military Operational
Settings: Download
the pdf file: Download
the pdf file: |
|
Introduction
What is Multiple Sclerosis? How Many People Have MS? Who Gets MS? How Much Does MS Cost America? What Causes MS? The Immune System Genetics What is the Course of MS? Can Life Events Affect the Course of MS? What are the Symptoms of MS? How is MS Diagnosed? Can MS be Treated? Immunotherapy Therapy to Improve Nerve Impulse Conduction Therapies Targeting an Antigen Cytokines Remyelination Diet Unproven Therapies Are Any MS Symptoms Treatable? What Recent Advances Have Been Made in MS Research? What Research Remains to be Done? What is the Outlook for People With MS? How Can I Help Research? Where can I get more information? Glossary IntroductionAlthough multiple sclerosis (MS) was first diagnosed in 1849, the earliest known description of a person with possible MS dates from fourteenth century Holland. An unpredictable disease of the central nervous system, MS can range from relatively benign to somewhat disabling to devastating as communication between the brain and other parts of the body is disrupted. The vast majority of patients are mildly affected, but in the worst cases MS can render a person unable to write, speak, or walk. A physician can diagnose MS in some patients soon after the onset of the illness. In others, however, physicians may not be able to readily identify the cause of the symptoms, leading to years of uncertainty and multiple diagnoses punctuated by baffling symptoms that mysteriously wax and wane. Once a diagnosis is made with confidence, patients must consider a profusion of information-and misinformation-associated with this complex disease. This brochure is designed to convey the latest information on the diagnosis, course, and possible treatment of MS, as well as highlights of current research. Although a pamphlet cannot substitute for the advice and expertise of a physician, it can provide patients and their families with information to understand MS better so that they can actively participate in their care and treatment. What is Multiple Sclerosis?During an MS attack, inflammation occurs in areas of the white matter* of the central nervous system in random patches called plaques. This process is followed by destruction of myelin, the fatty covering that insulates nerve cell fibers in the brain and spinal cord. Myelin facilitates the smooth, high-speed transmission of electrochemical messages between the brain, the spinal cord, and the rest of the body; when it is damaged, neurological transmission of messages may be slowed or blocked completely, leading to diminished or lost function. The name "multiple sclerosis" signifies both the number (multiple) and condition (sclerosis, from the Greek term for scarring or hardening) of the demyelinated areas in the central nervous system. How Many People Have MS?No one knows exactly how many people have MS. It is believed that, currently, there are approximately 250,000 to 350,000 people in the United States with MS diagnosed by a physician. This estimate suggests that approximately 200 new cases are diagnosed each week. Who Gets MS?Most people experience their first symptoms of MS between the ages of 20 and 40, but a diagnosis is often delayed. This is due to both the transitory nature of the disease and the lack of a specific diagnostic test-specific symptoms and changes in the brain must develop before the diagnosis is confirmed. Although scientists have documented cases of MS in young children and elderly adults, symptoms rarely begin before age 15 or after age 60. Whites are more than twice as likely as other races to develop MS. In general, women are affected at almost twice the rate of men; however, among patients who develop the symptoms of MS at a later age, the gender ratio is more balanced. MS is five times more prevalent in temperate climates-such as those found in the northern United States, Canada, and Europe-than in tropical regions. Furthermore, the age of 15 seems to be significant in terms of risk for developing the disease: some studies indicate that a person moving from a high-risk (temperate) to a low-risk (tropical) area before the age of 15 tends to adopt the risk (in this case, low) of the new area and vice versa. Other studies suggest that people moving after age 15 maintain the risk of the area where they grew up. These findings indicate a strong role for an environmental factor in the cause of MS. It is possible that, at the time of or immediately following puberty, patients acquire an infection with a long latency period. Or, conversely, people in some areas may come in contact with an unknown protective agent during the time before puberty. Other studies suggest that the unknown geographic or climatic element may actually be simply a matter of genetic predilection and reflect racial and ethnic susceptibility factors. Periodically, scientists receive reports of MS "clusters." The most famous of these MS "epidemics" took place in the Faeroe Islands north of Scotland in the years following the arrival of British troops during World War II. Despite intense study of this and other clusters, no direct environmental factor has been identified. Nor has any definitive evidence been found to link daily stress to MS attacks, although there is evidence that the risk of worsening is greater after acute viral illnesses. How Much Does MS Cost America?MS is a life-long chronic disease diagnosed primarily in young adults who have a virtually normal life expectancy. Consequently, the economic, social, and medical costs associated with the disease are significant. Estimates place the annual cost of MS in the United States in the billions of dollars. What Causes MS?Scientists have learned a great deal about MS in recent years; still, its cause remains elusive. Many investigators believe MS to be an autoimmune disease-one in which the body, through its immune system, launches a defensive attack against its own tissues. In the case of MS, it is the nerve-insulating myelin that comes under assault. Such assaults may be linked to an unknown environmental trigger, perhaps a virus. The Immune SystemTo understand what is happening when a person has MS, it is first necessary to know a little about how the healthy immune system works. The immune system - a complex network of specialized cells and organs - defends the body against attacks by "foreign" invaders such as bacteria, viruses, fungi, and parasites. It does this by seeking out and destroying the interlopers as they enter the body. Substances capable of triggering an immune response are called antigens. The immune system displays both enormous diversity and extraordinary specificity. It can recognize millions of distinctive foreign molecules and produce its own molecules and cells to match up with and counteract each of them. In order to have room for enough cells to match the millions of possible foreign invaders, the immune system stores just a few cells for each specific antigen. When an antigen appears, those few specifically matched cells are stimulated to multiply into a full-scale army. Later, to prevent this army from overexpanding, powerful mechanisms to suppress the immune response come into play. T cells, so named because they are processed in the thymus, appear to play a particularly important role in MS. They travel widely and continuously throughout the body patrolling for foreign invaders. In order to recognize and respond to each specific antigen, each T cell's surface carries special receptor molecules for particular antigens. T cells contribute to the body's defenses in two major ways. Regulatory T cells help orchestrate the elaborate immune system. For instance, they assist other cells to make antibodies, proteins programmed to match one specific antigen much as a key matches a lock. Antibodies typically interact with circulating antigens, such as bacteria, but are unable to penetrate living cells. Chief among the regulatory T cells are those known as helper (or inducer) cells. Helper T cells are essential for activating the body's defenses against foreign substances. Yet another subset of regulatory T cells acts to turn off, or suppress, various immune system cells when their job is done. Killer T cells, on the other hand, directly attack diseased or damaged body cells by binding to them and bombarding them with lethal chemicals called cytokines. Since T cells can attack cells directly, they must be able to discriminate between "self" cells (those of the body) and "nonself" cells (foreign invaders). To enable the immune system to distinguish the self, each body cell carries identifying molecules on its surface. T cells likely to react against the self are usually eliminated before leaving the thymus; the remaining T cells recognize the molecular markers and coexist peaceably with body tissues in a state of self-tolerance. In autoimmune diseases such as MS, the detente between the immune system and the body is disrupted when the immune system seems to wrongly identify self as nonself and declares war on the part of the body (myelin) it no longer recognizes. Through intensive research efforts, scientists are unraveling the complex secrets of the malfunctioning immune system of patients with MS. Components of myelin such as myelin basic protein have been the focus of much research because, when injected into laboratory animals, they can precipitate experimental allergic encephalomyelitis (EAE), a chronic relapsing brain and spinal cord disease that resembles MS. The injected myelin probably stimulates the immune system to produce anti-myelin T cells that attack the animal's own myelin. Investigators are also looking for abnormalities or malfunctions in the blood/brain barrier, a protective membrane that controls the passage of substances from the blood into the central nervous system. It is possible that, in MS, components of the immune system get through the barrier and cause nervous system damage. Scientists have studied a number of infectious agents (such as viruses) that have been suspected of causing MS, but have been unable to implicate any one particular agent. Viral infections are usually accompanied by inflammation and the production of gamma interferon, a naturally occurring body chemical that has been shown to worsen the clinical course of MS. It is possible that the immune response to viral infections may themselves precipitate an MS attack. There seems to be little doubt that something in the environment is involved in triggering MS. GeneticsIn addition, increasing scientific evidence suggests that genetics may play a role in determining a person's susceptibility to MS. Some populations, such as Gypsies, Eskimos, and Bantus, never get MS. Native Indians of North and South America, the Japanese, and other Asian peoples have very low incidence rates. It is unclear whether this is due mostly to genetic or environmental factors. In the population at large, the chance of developing MS is less than a tenth of one percent. However, if one person in a family has MS, that person's first-degree relatives-parents, children, and siblings-have a one to three percent chance of getting the disease. For identical twins, the likelihood that the second twin may develop MS if the first twin does is about 30 percent; for fraternal twins (who do not inherit identical gene pools), the likelihood is closer to that for non-twin siblings, or about 4 percent. The fact that the rate for identical twins both developing MS is significantly less than 100 percent suggests that the disease is not entirely genetically controlled. Some (but definitely not all) of this effect may be due to shared exposure to something in the environment, or to the fact that some people with MS lesions remain essentially asymptomatic throughout their lives. Further indications that more than one gene is involved in MS susceptibility comes from studies of families in which more than one member has MS. Several research teams found that people with MS inherit certain regions on individual genes more frequently than people without MS. Of particular interest is the human leukocyte antigen (HLA) or major histocompatibility complex region on chromosome 6. HLAs are genetically determined proteins that influence the immune system. The HLA patterns of MS patients tend to be different from those of people without the disease. Investigations in northern Europe and America have detected three HLAs that are more prevalent in people with MS than in the general population. Studies of American MS patients have shown that people with MS also tend to exhibit these HLAs in combination-that is, they have more than one of the three HLAs-more frequently than the rest of the population. Furthermore, there is evidence that different combinations of the HLAs may correspond to variations in disease severity and progression. Studies of families with multiple cases of MS and research comparing genetic regions of humans to those of mice with EAE suggest that another area related to MS susceptibility may be located on chromosome 5. Other regions on chromosomes 2, 3, 7, 11, 17, 19, and X have also been identified as possibly containing genes involved in the development of MS. These studies strengthen the theory that MS is the result of a number of factors rather than a single gene or other agent. Development of MS is likely to be influenced by the interactions of a number of genes, each of which (individually) has only a modest effect. Additional studies are needed to specifically pinpoint which genes are involved, determine their function, and learn how each gene's interactions with other genes and with the environment make an individual susceptible to MS. In addition to leading to better ways to diagnose MS, such studies should yield clues to the underlying causes of MS and, eventually, to better treatments or a way to prevent the disease. What is the Course of MS?Each case of MS displays one of several patterns of presentation and subsequent course. Most commonly, MS first manifests itself as a series of attacks followed by complete or partial remissions as symptoms mysteriously lessen, only to return later after a period of stability. This is called relapsing-remitting (RR) MS. Primary-progressive (PP) MS is characterized by a gradual clinical decline with no distinct remissions, although there may be temporary plateaus or minor relief from symptoms. Secondary-progressive (SP) MS begins with a relapsing-remitting course followed by a later primary-progressive course. Rarely, patients may have a progressive-relapsing (PR) course in which the disease takes a progressive path punctuated by acute attacks. PP, SP, and PR are sometimes lumped together and called chronic progressive MS. In addition, twenty percent of the MS population has a benign form of the disease in which symptoms show little or no progression after the initial attack; these patients remain fully functional. A few patients experience malignant MS, defined as a swift and relentless decline resulting in significant disability or even death shortly after disease onset. However, MS is very rarely fatal and most people with MS have a fairly normal life expectancy. Studies throughout the world are causing investigators to redefine the natural course of the disease. These studies use a technique called magnetic resonance imaging (MRI) to visualize the evolution of MS lesions in the white matter of the brain. Bright spots on a T2 MRI scan indicate the presence of lesions, but do not provide information about when they developed. Because investigators speculate that the breakdown of the blood/brain barrier is the first step in the development of MS lesions, it is important to distinguish new lesions from old. To do this, physicians give patients injections of gadolinium, a chemical contrast agent that normally does not cross the blood/brain barrier, before performing a scan. On this type of scan, called T1, the appearance of bright areas indicates periods of recent disease activity (when gadolinium is able to cross the barrier). The ability to estimate the age of lesions through MRI has allowed investigators to show that, in some patients, lesions occur frequently throughout the course of the disease even when no symptoms are present. Can Life Events Affect the Course of MS?While there is no good evidence that daily stress or trauma affects the course of MS, there is data on the influence of pregnancy. Since MS generally strikes during childbearing years, a common concern among women with the disease is whether or not to have a baby. Studies on the subject have shown that MS has no adverse effects on the course of pregnancy, labor, or delivery; in fact symptoms often stabilize or remit during pregnancy. This temporary improvement is thought to relate to changes in a woman's immune system that allow her body to carry a baby: because every fetus has genetic material from the father as well as the mother, the mother's body should identify the growing fetus as foreign tissue and try to reject it in much the same way the body seeks to reject a transplanted organ. To prevent this from happening, a natural process takes place to suppress the mother's immune system in the uterus during pregnancy. However, women with MS who are considering pregnancy need to be aware that certain drugs used to treat MS should be avoided during pregnancy and while breast feeding. These drugs can cause birth defects and can be passed to the fetus via blood and to an infant via breast milk. Among them are prednisone, corticotropin, azathioprine, cyclophosphamide, diazepam, phenytoin, carbamazepine, and baclofen. Unfortunately, between 20 and 40 percent of women with MS do have a relapse in the three months following delivery. However, there is no evidence that pregnancy and childbirth affect the overall course of the disease one way or the other. Also, while MS is not in itself a reason to avoid pregnancy and poses no significant risks to the fetus, physical limitations can make child care more difficult. It is therefore important that MS patients planning families discuss these issues with both their partner and physician. What are the Symptoms of MS?Symptoms of MS may be mild or severe, of long duration or short, and may appear in various combinations, depending on the area of the nervous system affected. Complete or partial remission of symptoms, especially in the early stages of the disease, occurs in approximately 70 percent of MS patients. The initial symptom of MS is often blurred or double vision, red-green color distortion, or even blindness in one eye. Inexplicably, visual problems tend to clear up in the later stages of MS. Inflammatory problems of the optic nerve may be diagnosed as retrobulbaror optic neuritis. Fifty-five percent of MS patients will have an attack of optic neuritis at some time or other and it will be the first symptom of MS in approximately 15 percent. This has led to general recognition of optic neuritis as an early sign of MS, especially if tests also reveal abnormalities in the patient's spinal fluid. Most MS patients experience muscle weakness in their extremities and difficulty with coordination and balance at some time during the course of the disease. These symptoms may be severe enough to impair walking or even standing. In the worst cases, MS can produce partial or complete paralysis. Spasticity-the involuntary increased tone of muscles leading to stiffness and spasms-is common, as is fatigue. Fatigue may be triggered by physical exertion and improve with rest, or it may take the form of a constant and persistent tiredness. Most people with MS also exhibit paresthesias, transitory abnormal sensory feelings such as numbness, prickling, or "pins and needles" sensations; uncommonly, some may also experience pain. Loss of sensation sometimes occurs. Speech impediments, tremors, and dizziness are other frequent complaints. Occasionally, people with MS have hearing loss. Approximately half of all people with MS experience cognitive impairments such as difficulties with concentration, attention, memory, and poor judgment, but such symptoms are usually mild and are frequently overlooked. In fact, they are often detectable only through comprehensive testing. Patients themselves may be unaware of their cognitive loss; it is often a family member or friend who first notices a deficit. Such impairments are usually mild, rarely disabling, and intellectual and language abilities are generally spared. Cognitive symptoms occur when lesions develop in brain areas responsible for information processing. These deficits tend to become more apparent as the information to be processed becomes more complex. Fatigue may also add to processing difficulties. Scientists do not yet know whether altered cognition in MS reflects problems with information acquisition, retrieval, or a combination of both. Types of memory problems may differ depending on the individual's disease course (relapsing-remitting, primary-progressive, etc.), but there does not appear to be any direct correlation between duration of illness and severity of cognitive dysfunction. . Depression, which is unrelated to cognitive problems, is another common feature of MS. In addition, about 10 percent of patients suffer from more severe psychotic disorders such as manic-depression and paranoia. Five percent may experience episodes of inappropriate euphoria and despair-unrelated to the patient's actual emotional state-known as "laughing/weeping syndrome." This syndrome is thought to be due to demyelination in the brainstem, the area of the brain that controls facial expression and emotions, and is usually seen only in severe cases. As the disease progresses, sexual dysfunction may become a problem. Bowel and bladder control may also be lost. In about 60 percent of MS patients, heat-whether generated by temperatures outside the body or by exercise-may cause temporary worsening of many MS symptoms. In these cases, eradicating the heat eliminates the problem. Some temperature-sensitive patients find that a cold bath may temporarily relieve their symptoms. For the same reason, swimming is often a good exercise choice for people with MS. The erratic symptoms of MS can affect the entire family as patients may become unable to work at the same time they are facing high medical bills and additional expenses for housekeeping assistance and modifications to homes and vehicles. The emotional drain on both patient and family is immeasurable. Support groups (listed on a card in the pocket at the back of this pamphlet) and counseling may help MS patients, their families, and friends find ways to cope with the many problems the disease can cause. Possible Symptoms of Multiple Sclerosis
How is MS Diagnosed?There is no single test that unequivocally detects MS. When faced with a patient whose symptoms, neurological exam results, and medical history suggest MS, physicians use a variety of tools to rule out other possible disorders and perform a series of laboratory tests which, if positive, confirm the diagnosis. Imaging technologies such as MRI can help locate central nervous system lesions resulting from myelin loss. MRI is painless, noninvasive, and does not expose the body to radiation. It is often used in conjunction with the contrast agent gadolinium, which helps distinguish new plaques from old. However, since these lesions can also occur in several other neurological disorders, they are not absolute evidence of MS. Several new MRI techniques may help quantify and characterize MS lesions that are too subtle to be detected using conventional MRI scans. While standard MRI provides an anatomical picture of lesions, magnetic resonance spectroscopy (MRS) yields information about the brain's biochemistry; specifically, it can measure the brain chemical N-acetyl aspartate. Decreased levels of this chemical can indicate nerve damage. Magnetization transfer imaging (MTI) is able to detect white matter abnormalities before lesions can be seen on standard MRI scans by calculating the amount of "free" water in tissues. Demyelinated tissues and damaged nerves show increased levels of free" (versus "bound") water particles. Diffusion-tensor magnetic resonance imaging (DT-MRI or DTI) measures the random motion of water molecules. Individual water molecules are constantly in motion, colliding with each other at extremely high speeds. This causes them to spread out, or diffuse. DT-MRI maps this diffusion to produce intricate, three-dimensional images indicating the size and location of demyelinated areas of the brain. Changes in this process can then be measured and correlated with disease progression. Functional MRI (fMRI) uses radio waves and a strong magnetic field to measures the correlation between physical changes in the brain (such as blood flow) and mental functioning during the performance of cognitive tasks. In addition to helping scientists and physicians better understand how MS develops-an important first step in devising new treatments-these approaches offer earlier diagnosis and enhance efforts to monitor disease progression and the effects of treatment. Other tests that may be used to diagnosis MS include visual evoked potential (VEP) tests and studies of cerebrospinal fluid (the colorless liquid that circulates through the brain and spinal cord). VEP tests measure the speed of the brain's response to visual stimuli. VEP can sometimes detect lesions that the scanners miss and is particularly useful when abnormalities seen on MRI do not meet the specific criteria for MS. Auditory and sensory evoked potentials have also been used in the past, but are no longer believed to contribute significantly to the diagnosis of MS. Like imaging technologies, VEP is helpful but not conclusive because it cannot identify the cause of lesions. Examination of cerebrospinal fluid can show cellular and chemical abnormalities often associated with MS. These abnormalities include increased numbers of white blood cells and higher-than-average amounts of protein, especially myelin basic protein and an antibody called immunoglobulin G. Physicians can use several different laboratory techniques to separate and graph the various proteins in MS patients' cerebrospinal fluid. This process often identifies the presence of a characteristic pattern called oligoclonal bands. While it can still be difficult for the physician to differentiate between an MS attack and symptoms that can follow a viral infection or even an immunization, our growing understanding of disease mechanisms and the expanded use of MRI is enabling physicians to diagnose MS with far more confidence than ever before. Today, most patients who undergo a diagnostic evaluation for MS will be classified as either having MS or not having MS, although there are still cases where a person may have the clinical symptoms of MS but not meet all the criteria to confirm a diagnosis of MS. In these cases, a diagnosis of "possible MS" is used. A number of other diseases may produce symptoms similar to those seen in MS. Other conditions with an intermittent course and MS-like lesions of the brain's white matter include polyarteritis, lupus erythematosus, syringomyelia, tropical spastic paraparesis, some cancers, and certain tumors that compress the brainstem or spinal cord. Progressive multifocal leukoencephalopathy can mimic the acute stage of an MS attack. Physicians will also need to rule out stroke, neurosyphilis, spinocerebellar ataxias, pernicious anemia, diabetes, Sjogren's disease, and vitamin B12 deficiency. Acute transverse myelitis may signal the first attack of MS, or it may indicate other problems such as infection with the Epstein-Barr or herpes simplex B viruses. Recent reports suggest that the neurological problems associated with Lyme disease may present a clinical picture much like MS. Investigators are continuing their search for a definitive test for MS. Until one is developed, however, evidence of both multiple attacks and central nervous system lesions must be found before a diagnosis of MS is given. Can MS be Treated?There is as yet no cure for MS. Many patients do well with no therapy at all, especially since many medications have serious side effects and some carry significant risks. Naturally occurring or spontaneous remissions make it difficult to determine therapeutic effects of experimental treatments; however, the emerging evidence that MRIs can chart the development of lesions is already helping scientists evaluate new therapies. In the past, the principal medications physicians used to treat MS were steroids possessing anti-inflammatory properties; these include adrenocorticotropic hormone (better known as ACTH), prednisone, prednisolone, methylprednisolone, betamethasone, and dexamethasone. Studies suggest that intravenous methylprednisolone may be superior to the more traditional intravenous ACTH for patients experiencing acute relapses; no strong evidence exists to support the use of these drugs to treat progressive forms of MS. Also, there is some indication that steroids may be more appropriate for people with movement, rather than sensory, symptoms. While steroids do not affect the course of MS over time, they can reduce the duration and severity of attacks in some patients. The mechanism behind this effect is not known; one study suggests the medications work by restoring the effectiveness of the blood/brain barrier. Because steroids can produce numerous adverse side effects (acne, weight gain, seizures, psychosis), they are not recommended for long-term use. One of the most promising MS research areas involves naturally occurring antiviral proteins known as interferons. Three forms of beta interferon (Avonex, Betaseron, and Rebif) have now been approved by the Food and Drug Administration for treatment of relapsing-remitting MS. Beta interferon has been shown to reduce the number of exacerbations and may slow the progression of physical disability. When attacks do occur, they tend to be shorter and less severe. In addition, MRI scans suggest that beta interferon can decrease myelin destruction. Investigators speculate that the effects of beta interferon may be due to the drug's ability to correct an MS-related deficiency of certain white blood cells that suppress the immune system and/or its ability to inhibit gamma interferon, a substance believed to be involved in MS attacks. Alpha interferon is also being studied as a possible treatment for MS. Common side effects of interferons include fever, chills, sweating, muscle aches, fatigue, depression, and injection site reactions. Scientists continue their extensive efforts to create new and better therapies for MS. Goals of therapy are threefold: to improve recovery from attacks, to prevent or lessen the number of relapses, and to halt disease progression. Some therapies currently under investigation are discussed below. ImmunotherapyAs evidence of immune system involvement in the development of MS has grown, trials of various new treatments to alter or suppress immune response are being conducted. Most of these therapies are, at this time, still considered experimental. Results of recent clinical trials have shown that immunosuppressive agents and techniques can positively (if temporarily) affect the course of MS; however, toxic side effects often preclude their widespread use. In addition, generalized immunosuppression leaves the patient open to a variety of viral, bacterial, and fungal infections. Over the years, MS investigators have studied a number of immunosuppressant treatments. One such treatment, Novantrone (mitoxantrone), was approved by the FDA for the treatment of advanced or chronic MS. Other therapies being studied are cyclosporine (Sandimmune), cyclophosphamide (Cytoxan), methotrexate, azathioprine (Imuran), and total lymphoid irradiation (a process whereby the MS patient's lymph nodes are irradiated with x-rays in small doses over a few weeks to destroy lymphoid tissue, which is actively involved in tissue destruction in autoimmune diseases). Inconclusive and/or contradictory results of these trials, combined with the therapies' potentially dangerous side effects, dictate that further research is necessary to determine what, if any, role they should play in the management of MS. Studies are also being conducted with the immune system modulating drug cladribine (Leustatin). Another potential treatment for MS is monoclonal antibodies, which are identical, laboratory-produced antibodies that are highly specific for a single antigen. They are injected into the patient in the hope that they will alter the patient's immune response. One monoclonal antibody, natalizumab (Tysabri), was shown in clinical trials to significantly reduce the frequency of attacks in people with relapsing forms of MS and was approved for marketing by the U.S. Food and Drug Administration (FDA) in 2004. However, in 2005 the drug’s manufacturer voluntarily suspended marketing of the drug after several reports of significant adverse events. In 2006, the FDA again approved sale of the drug for MS but under strict treatment guidelines involving infusion centers where patients can be monitored by specially trained physicians. Another experimental treatment for MS is plasma exchange, or plasmapheresis. Plasmapheresis is a procedure in which blood is removed from the patient and the blood plasma is separated from other blood substances that may contain antibodies and other immunologically active products. These other blood substances are discarded and the plasma is then transfused back into the patient. Because its worth as a treatment for MS has not yet been proven, this experimental treatment remains at the stage of clinical testing. Bone marrow transplantation (a procedure in which bone marrow from a healthy donor is infused into patients who have undergone drug or radiation therapy to suppress their immune system so they will not reject the donated marrow) and injections of venom from honey bees are also being studied. Each of these therapies carries the risk of potentially severe side effects. Therapy to Improve Nerve Impulse ConductionBecause the transmission of electrochemical messages between the brain and body is disrupted in MS, medications to improve the conduction of nerve impulses are being investigated. Since demyelinated nerves show abnormalities of potassium activity, scientists are studying drugs that block the channels through which potassium moves, thereby restoring conduction of the nerve impulse. In several small experimental trials, derivatives of a drug called aminopyridine temporarily improved vision, coordination, and strength when given to MS patients who suffered from both visual symptoms and heightened sensitivity to temperature. Possible side effects of these therapies include paresthesias (tingling sensations), dizziness, and seizures. Therapies Targeting an AntigenTrials of a synthetic form of myelin basic protein, called copolymer I (Copaxone), were successful, leading the FDA to approve the agent for the treatmernt of relapsing-remitting MS. Copolymer I, unlike so many drugs tested for the treatment of MS, has few side effects, and studies indicate that the agent can reduce the relapse rate by almost one third. In addition, patients given copolymer I are more likely to show neurologic improvement than those given a placebo. Investigators are also looking at the possibility of developing an MS vaccine. Myelin-attacking T cells were removed, inactivated, and injected back into animals with experimental allergic encephalomyelitis (EAE). This procedure results in destruction of the immune system cells that were attacking myelin basic protein. In a couple of small trials scientists have tested a similar vaccine in humans. The product was well-tolerated and had no side effects, but the studies were too small to establish efficacy. Patients with progressive forms of MS did not appear to benefit, although relapsing-remitting patients showed some neurologic improvement and had fewer relapses and reduced numbers of lesions in one study. Unfortunately, the benefits did not last beyond two years. A similar approach, known as peptide therapy, is based on evidence that the body can mount an immune response against the T cells that destroy myelin, but this response is not strong enough to overcome the disease. To induce this response, the investigator scans the myelin-attacking T cells for the myelin-recognizing receptors on the cells' surface. A fragment, or peptide, of those receptors is then injected into the body. The immune system "sees" the injected peptide as a foreign invader and launches an attack on any myelin-destroying T cells that carry the peptide. The injection of portions of T cell receptors may heighten the immune system reaction against the errant T cells much the same way a booster shot heightens immunity to tetanus. Or, peptide therapy may jam the errant cells' receptors, preventing the cells from attacking myelin. Despite these promising early results, there are some major obstacles to developing vaccine and peptide therapies. Individual patients' T cells vary so much that it may not be possible to develop a standard vaccine or peptide therapy beneficial to all, or even most, MS patients. At this time, each treatment involves extracting cells from each individual patient, purifying the cells, and then growing them in culture before inactivating and chemically altering them. This makes the production of quantities sufficient for therapy extremely time consuming, labor intensive, and expensive. Further studies are necessary to determine whether universal inoculations can be developed to induce suppression of MS patients' overactive immune systems. Protein antigen feeding is similar to peptide therapy, but is a potentially simpler means to the same end. Whenever we eat, the digestive system breaks each food or substance into its primary "non-antigenic" building blocks, thereby averting a potentially harmful immune attack. So, strange as it may seem, antigens that trigger an immune response when they are injected can encourage immune system tolerance when taken orally. Furthermore, this reaction is directed solely at the specific antigen being fed; wholesale immunosuppression, which can leave the body open to a variety of infections, does not occur. Studies have shown that when rodents with EAE are fed myelin protein antigens, they experience fewer relapses. Data from a small, preliminary trial of antigen feeding in humans found limited suggestion of improvement, but the results were not statistically significant. A multi-center trial is being conducted to determine whether protein antigen feeding is effective. CytokinesAs our growing insight into the workings of the immune system gives us new knowledge about the function of cytokines, the powerful chemicals produced by T cells, the possibility of using them to manipulate the immune system becomes more attractive. Scientists are studying a variety of substances that may block harmful cytokines, such as those involved in inflammation, or that encourage the production of protective cytokines. A drug that has been tested as a depression treatment, rolipram, has been shown to reduce levels of several destructive cytokines in animal models of MS. Its potential as a therapy for MS is not known at this time, but side effects seem modest. Protein antigen feeding, discussed above, may release transforming growth factor beta (TGF), a protective cytokine that inhibits or regulates the activity of certain immune cells. Preliminary tests indicate that it may reduce the number of immune cells commonly found in MS patients' spinal fluid. Side effects include anemia and altered kidney function. Interleukin 4 (IL-4) is able to diminish demyelination and improve the clinical course of mice with EAE, apparently by influencing developing T cells to become protective rather than harmful. This also appears to be true of a group of chemicals called retinoids. When fed to rodents with EAE, retinoids increase levels of TGF and IL-4, which encourage protective T cells, while decreasing numbers of harmful T cells. This results in improvement of the animals' clinical symptoms. RemyelinationSome studies focus on strategies to reverse the damage to myelin and oligodendrocytes (the cells that make and maintain myelin in the central nervous system), both of which are destroyed during MS attacks. Scientists now know that oligodendrocytes may proliferate and form new myelin after an attack. Therefore, there is a great deal of interest in agents that may stimulate this reaction. To learn more about the process, investigators are looking at how drugs used in MS trials affect remyelination. Studies of animal models indicate that monoclonal antibodies and two immunosuppressant drugs, cyclophosphamide and azathioprine, may accelerate remyelination, while steroids may inhibit it. The ability of intravenous immunoglobulin (IVIg) to restore visual acuity and/or muscle strength is also being investigated. DietOver the years, many people have tried to implicate diet as a cause of or treatment for MS. Some physicians have advocated a diet low in saturated fats; others have suggested increasing the patient's intake of linoleic acid, a polyunsaturated fat, via supplements of sunflower seed, safflower, or evening primrose oils. Other proposed dietary "remedies" include megavitamin therapy, including increased intake of vitamins B12 or C; various liquid diets; and sucrose-, tobacco-, or gluten-free diets. To date, clinical studies have not been able to confirm benefits from dietary changes; in the absence of any evidence that diet therapy is effective, patients are best advised to eat a balanced, wholesome diet. Unproven TherapiesMS is a disease with a natural tendency to remit spontaneously, and for which there is no universally effective treatment and no known cause. These factors open the door for an array of unsubstantiated claims of cures. At one time or another, many ineffective and even potentially dangerous therapies have been promoted as treatments for MS. A partial list of these "therapies" includes: injections of snake venom, electrical stimulation of the spinal cord's dorsal column, removal of the thymus gland, breathing pressurized (hyperbaric) oxygen in a special chamber, injections of beef heart and hog pancreas extracts, intravenous or oral calcium orotate (calcium EAP), hysterectomy, removal of dental fillings containing silver or mercury amalgams, and surgical implantation of pig brain into the patient's abdomen. None of these treatments is an effective therapy for MS or any of its symptoms. Are Any MS Symptoms Treatable?While some scientists look for therapies that will affect the overall course of the disease, others are searching for new and better medications to control the symptoms of MS without triggering intolerable side effects. Many people with MS have problems with spasticity, a condition that primarily affects the lower limbs. Spasticity can occur either as a sustained stiffness caused by increased muscle tone or as spasms that come and go, especially at night. It is usually treated with muscle relaxants and tranquilizers. Baclofen (Lioresal), the most commonly prescribed medication for this symptom, may be taken orally or, in severe cases, injected into the spinal cord. Tizanidine (Zanaflex), used for years in Europe and now approved in the United States, appears to function similarly to baclofen. Diazepam (Valium), clonazepam (Klonopin), and dantrolene (Dantrium) can also reduce spasticity. Although its beneficial effect is temporary, physical therapy may also be useful and can help prevent the irreversible shortening of muscles known as contractures. Surgery to reduce spasticity is rarely appropriate in MS. Weakness and ataxia (incoordination) are also characteristic of MS. When weakness is a problem, some spasticity can actually be beneficial by lending support to weak limbs. In such cases, medication levels that alleviate spasticity completely may be inappropriate. Physical therapy and exercise can also help preserve remaining function, and patients may find that various aids-such as foot braces, canes, and walkers-can help them remain independent and mobile. Occasionally, physicians can provide temporary relief from weakness, spasms, and pain by injecting a drug called phenol into the spinal cord, muscles, or nerves in the arms or legs. Further research is needed to find or develop effective treatments for MS-related weakness and ataxia. Although improvement of optic symptoms usually occurs even without treatment, a short course of treatment with intravenous methylprednisolone (Solu-Medrol) followed by treatment with oral steroids is sometimes used. A trial of oral prednisone in patients with visual problems suggests that this steroid is not only ineffective in speeding recovery but may also increase patients' risk for future MS attacks. Curiously, prednisone injected directly into the veins-at ten times the oral dose-did seem to produce short-term recovery. Because of the link between optic neuritis and MS, the study's investigators believe these findings may hold true for the treatment of MS as well. A follow-up study of optic neuritis patients will address this and other questions. Fatigue, especially in the legs, is a common symptom of MS and may be both physical and psychological. Avoiding excessive activity and heat are probably the most important measures patients can take to counter physiological fatigue. If psychological aspects of fatigue such as depression or apathy are evident, antidepressant medications may help. Other drugs that may reduce fatigue in some, but not all, patients include amantadine (Symmetrel), pemoline (Cylert), and the still-experimental drug aminopyridine. People with MS may experience several types of pain. Muscle and back pain can be helped by aspirin or acetaminophen and physical therapy to correct faulty posture and strengthen and stretch muscles. The sharp, stabbing facial pain known as trigeminal neuralgia is commonly treated with carbamazepine or other anticonvulsant drugs or, occasionally, surgery. Intense tingling and burning sensations are harder to treat. Some people get relief with antidepressant drugs; others may respond to electrical stimulation of the nerves in the affected area. In some cases, the physician may recommend codeine. As the disease progresses, some patients develop bladder malfunctions. Urinary problems are often the result of infections that can be treated with antibiotics. The physician may recommend that patients take vitamin C supplements or drink cranberry juice, as these measures acidify urine and may reduce the risk of further infections. Several medications are also available. The most common bladder problems encountered by MS patients are urinary frequency, urgency, or incontinence. A small number of patients, however, retain large amounts of urine. In these patients, catheterization may be necessary. In this procedure, a catheter or drainage tube is temporarily inserted (by the patient or a caretaker) into the urethra several times a day to drain urine from the bladder. Surgery may be indicated in severe, intractable cases. Scientists have developed a "bladder pacemaker" that has helped people with urinary incontinence in preliminary trials. The pacemaker, which is surgically implanted, is controlled by a hand-held unit that allows the patient to electrically stimulate the nerves that control bladder function. MS patients with urinary problems may be reluctant to drink enough fluids, leading to constipation. Drinking more water and adding fiber to the diet usually alleviates this condition. Sexual dysfunction may also occur, especially in patients with urinary problems. Men may experience occasional failure to attain an erection. Penile implants, injection of the drug papaverine, and electrostimulation are techniques used to resolve the problem. Women may experience insufficient lubrication or have difficulty reaching orgasm; in these cases, vaginal gels and vibrating devices may be helpful. Counseling is also beneficial, especially in the absence of urinary problems, since psychological factors can also cause these symptoms. For instance, depression can intensify symptoms of fatigue, pain, and sexual dysfunction. In addition to counseling, the physician may prescribe antidepressant or antianxiety medications. Amitriptyline is used to treat laughing/weeping syndrome. Tremors are often resistant to therapy, but can sometimes be treated with drugs or, in extreme cases, surgery. Investigators are currently examining a number of experimental treatments for tremor. Drugs Used to Treat Symptoms of Multiple Sclerosis
What Recent Advances Have Been Made in MS Research?Many advances, on several fronts, have been made in the war against MS. Each advance interacts with the others, adding greater depth and meaning to each new discovery. Four areas, in particular, stand out. Over the last decade, our knowledge about how the immune system works has grown at an amazing rate. Major gains have been made in recognizing and defining the role of this system in the development of MS lesions, giving scientists the ability to devise ways to alter the immune response. Such work is expected to yield a variety of new potential therapies that may ameliorate MS without harmful side effects. New tools such as MRI have redefined the natural history of MS and are proving invaluable in monitoring disease activity. Scientists are now able to visualize and follow the development of MS lesions in the brain and spinal cord using MRI; this ability is a tremendous aid in the assessment of new therapies and can speed the process of evaluating new treatments. Other tools have been developed that make the painstaking work of teasing out the disease's genetic secrets possible. Such studies have strengthened scientists' conviction that MS is a disease with many genetic components, none of which is dominant. Immune system-related genetic factors that predispose an individual to the development of MS have been identified, and may lead to new ways to treat or prevent the disease. In fact, a treatment that may actually slow the course of the disease has been found and a growing number of therapies are now available that effectively treat some MS symptoms. In addition, there are a number of treatments under investigation that may curtail attacks or improve function of demyelinated nerve fibers. Over a dozen clinical trials testing potential therapies are under way, and additional new treatments are being devised and tested in animal models. What Research Remains to be Done?The role of genetic risk factors, and how they can be modified, must be more clearly defined. Environmental triggers, such as viruses or toxins, need to be investigated further. The specific cellular and subcellular targets of immune attack in the brain and spinal cord, and the subsets of T cells involved in that attack, need to be identified. Knowledge of these aspects of the disease will enable scientists to develop new methods for halting-or reversing and repairing-the destruction of myelin that causes the symptoms of MS. What is the Outlook for People With MS?The 1990s-proclaimed the "Decade of the Brain" in 1989 by President Bush and Congress-have seen an unparalleled explosion of knowledge about neurological disorders. New technologies are forcing even complex diseases like MS to yield up their secrets. These burgeoning opportunities in the field of neurological research have prompted the National Advisory Neurological Disorders and Stroke Council to suggest that an effective treatment for and the cause of MS may be found during the Decade of the Brain. The former has already been achieved; scientists continue to diligently search for the latter. Their dedication is the best hope for a cure, or, better yet, a way to prevent MS altogether. How Can I Help Research?The NINDS contributes to the support of the Human Brain and Spinal Fluid Resource Center in Los Angeles. This bank supplies investigators around the world with tissue from patients with neurological and other disorders. Tissue from individuals with MS is needed to enable scientists to study this disorder more intensely. Prospective donors may contact: Human Brain and Spinal Fluid Resource Center Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
antibodies - proteins made by the immune system that bind to structures (antigens) they recognize as foreign to the body. antigen - a structure foreign to the body, such as a virus. The body usually responds to antigens by producing antibodies. ataxia - a condition in which the muscles fail to function in a coordinated manner. autoimmune disease - a disease in which the body's defense system malfunctions and attacks a part of the body itself rather than foreign matter. blood/brain barrier - a membrane that controls the passage of substances from the blood into the central nervous system. cerebrospinal fluid - the colorless liquid, consisting partially of substances filtered from blood and partially by secretions released by brain cells, that circulates around and through the cavities of the brain and spinal cord. Physicians use a variety of tests-electrophoresis, isoelectric focusing, capillary isotachophoresis, and radioimmunoassay-to study cerebrospinal fluid for abnormalities often associated with MS. cytokines - powerful chemical substances secreted by T cells. Cytokines are an important factor in the production of inflammation and show promise as treatments for MS. demyelination - damage caused to myelin by recurrent attacks of inflammation. Demyelination ultimately results in nervous system scars, called plaques, which interrupt communications between the nerves and the rest of the body. experimental allergic encephalomyelitis (EAE) - a chronic brain and spinal cord disease similar to MS which is induced by injecting myelin basic protein into laboratory animals. fatigue - tiredness that may accompany activity or may persist even without exertion. gadolinium - a chemical compound given during MRI scans that helps distinguish new lesions from old. human leukocyte antigens (HLAs) - antigens, tolerated by the body, that correspond to genes that govern immune responses. Also known as major histocompatibility complex.immunoglobulin G (IgG) - an antibody-containing substance produced by human plasma cells in diseased central nervous system plaques. Levels of IgG are increased in the cerebrospinal fluid of most MS patients. immunosuppression - suppression of immune system functions. Many medications under investigation for the treatment of MS are immunosuppressants. interferons - cytokines belonging to a family of antiviral proteins that occur naturally in the body. Gamma interferon is produced by immune system cells, enhances T-cell recognition of antigens, and causes worsening of MS symptoms. Alpha and beta interferon probably exert a suppressive effect on the immune system and may be beneficial in the treatment of MS. lesion - an abnormal change in the structure of an organ due to disease or injury. magnetic resonance imaging (MRI) - a non-invasive scanning technique that enables investigators to see and track MS lesions as they evolve. myelin - a fatty covering insulating nerve cell fibers in the brain and spinal cord, myelin facilitates the smooth, high-speed transmission of electrochemical messages between these components of the central nervous system and the rest of the body. In MS, myelin is damaged through a process known as demyelination, which results in distorted or blocked signals. myelin basic protein (MBP) - a major component of myelin. When myelin breakdown occurs (as in MS), MBP can often be found in abnormally high levels in the patient's cerebrospinal fluid. When injected into laboratory animals, MBP induces experimental allergic encephalomyelitis, a chronic brain and spinal cord disease similar to MS. oligodendrocytes - cells that make and maintain myelin. optic neuritis - an inflammatory disorder of the optic nerve that usually occurs in only one eye and causes visual loss and sometimes blindness. It is generally temporary. paresthesias - abnormal sensations such as numbness, prickling, or "pins and needles." plaques - patchy areas of inflammation and demyelination typical of MS, plaques disrupt or block nerve signals that would normally pass through the regions affected by the plaques. receptor - a protein on a cell's surface that allows the cell to identify antigens. retrobulbar neuritis - an inflammatory disorder of the optic nerve that is usually temporary. It causes rapid loss of vision and may cause pain upon moving the eye. spasticity - involuntary muscle contractions leading to spasms and stiffness or rigidity. In MS, this condition primarily affects the lower limbs. T cells - immune system cells that develop in the thymus gland. Findings suggest that T cells are implicated in myelin destruction. transverse myelitis - an acute spinal cord disorder causing sudden low back pain and muscle weakness and abnormal sensory sensations in the lower extremities. Transverse myelitis often remits spontaneously; however, severe or long-lasting cases may lead to permanent disability. white matter - nerve fibers that are the site of MS lesions and underlie the gray matter of the brain and spinal cord. NIH Publication No. 96-75
|
|
Introduction
What is muscular dystrophy? What causes MD? How many people have MD? How does MD affect muscles? Are there other MD-like conditions? How do the muscular dystrophies differ? How are the muscular dystrophies diagnosed? How are the muscular dystrophies treated? What is the prognosis? What research is being done? Where can I get more information? Glossary IntroductionThe first historical account of muscular dystrophy appeared in 1830, when Sir Charles Bell wrote an essay about an illness that caused progressive weakness in boys. Six years later, another scientist reported on two brothers who developed generalized weakness, muscle damage, and replacement of damaged muscle tissue with fat and connective tissue. At that time the symptoms were thought to be signs of tuberculosis. In the 1850s, descriptions of boys who grew progressively weaker, lost the ability to walk, and died at an early age became more prominent in medical journals. In the following decade, French neurologist Guillaume Duchenne gave a comprehensive account of 13 boys with the most common and severe form of the disease (which now carries his name-Duchenne muscular dystrophy). It soon became evident that the disease had more than one form, and that these diseases affected people of either sex and of all ages. What is muscular dystrophy?Muscular dystrophy (MD) refers to a group of more than 30 genetic diseases that cause progressive weakness and degeneration of skeletal muscles used during voluntary movement. The word dystrophy is derived from the Greek dys, which means "difficult" or "faulty," and troph, or "nourish." These disorders vary in age of onset, severity, and pattern of affected muscles. All forms of MD grow worse as muscles progressively degenerate and weaken. The majority of patients eventually lose the ability to walk. Some types of MD also affect the heart, gastrointestinal system, endocrine glands, spine, eyes, brain, and other organs. Respiratory and cardiac diseases are common, and some patients may develop a swallowing disorder. MD is not contagious and cannot be brought on by injury or activity. What causes MD?All of the muscular dystrophies are inherited and involve a mutation in one of the thousands of genes that program proteins critical to muscle integrity. The body's cells don't work properly when a protein is altered or produced in insufficient quantity (or sometimes missing completely). Many cases of MD occur from spontaneous mutations that are not found in the genes of either parent, and this defect can be passed to the next generation. Genes are like blueprints: they contain coded messages that determine a person's characteristics or traits. They are arranged along 23 rod-like pairs of chromosomes, * with one half of each pair being inherited from each parent. Each half of a chromosome pair is similar to the other, except for one pair, which determines the sex of the individual. Muscular dystrophies can be inherited in three ways:
*Terms in Italics are defined in the glossary. How many people have MD?MD occurs worldwide, affecting all races. Its incidence varies, as some forms are more common than others. Its most common forms in children, Duchenne and Becker muscular dystrophy, alone affect approximately 1 in every 3,500 to 5,000 boys, or between 400 and 600 live male births each year in the United States.** Some types of MD are more prevalent in certain countries and regions of the world. Most muscular dystrophies are familial, meaning there is some family history of the disease. ** Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities, July 27, 2005 How does MD affect muscles?Muscles are made up of thousands of muscle fibers. Each fiber is actually a number of individual cells that have joined together during development and are encased by an outer membrane. Muscle fibers that make up individual muscles are bound together by connective tissue. Muscles are activated when an impulse, or signal, is sent from the brain along the peripheral nerves (nerves that connect the central nervous system to sensory organs and muscles) to the neuromuscular junction (the space between the nerve fiber and the muscle it activates). There, a release of the chemical acetylcholine triggers a series of events that cause the muscle to contract. The muscle fiber membrane contains a group of proteins-called the dystrophin-glycoprotein complex-which prevents damage as muscle fibers contract and relax. When this protective membrane is damaged, muscle fibers begin to leak the protein creatine kinase (needed for the chemical reactions that produce energy for muscle contractions) and take on excess calcium, which causes further harm. Affected muscle fibers eventually die from this damage, leading to progressive muscle degeneration. Although MD can affect several body tissues and organs, it most prominently affects the integrity of muscle fibers. The disease causes muscle degeneration, progressive weakness, fiber death, fiber branching and splitting, phagocytosis (in which muscle fiber material is broken down and destroyed by scavenger cells), and, in some cases, chronic or permanent shortening of tendons and muscles. Also, overall muscle strength and tendon reflexes are usually lessened or lost due to replacement of muscle by connective tissue and fat. Are there other MD-like conditions?There are many other heritable diseases that affect the muscles, the nerves, or the neuromuscular junction. These diseases may produce symptoms that are very similar to those found in some forms of MD (such as inflammatory myopathy, progressive muscle weakness, mental impairment, and cardiomyopathy) but they are caused by different genetic defects. The sharing of symptoms among multiple neuromuscular diseases, and the prevalence of sporadic cases in families not previously affected by MD, often makes it difficult for MD patients to obtain a quick diagnosis. Studies of other related muscle diseases may, however, contribute to what we know about MD. How do the muscular dystrophies differ?There are nine major groups of the muscular dystrophies. The disorders are classified by the extent and distribution of muscle weakness, age of onset, rate of progression, severity of symptoms, and family history (including any pattern of inheritance). Although some forms of MD become apparent in infancy or childhood, others may not appear until middle age or later. Overall, incidence rates and severity vary, but each of the dystrophies causes progressive skeletal muscle deterioration, and some types affect cardiac muscle. There are four forms of MD that begin in childhood: Duchenne MD is the most common childhood form of MD, as well as the most common of the muscular dystrophies overall, accounting for approximately 50 percent of all cases. It affects approximately one in 3,500 male births. Because inheritance is X-linked recessive (caused by a mutation on the X, or sex, chromosome), Duchenne MD primarily affects boys, although girls and women who carry the defective gene may show some symptoms. About one-third of the cases reflect new mutations and the rest run in families. Sisters of boys with Duchenne MD have a 50 percent chance of carrying the defective gene. Duchenne MD usually becomes apparent when an affected child begins to walk. Progressive weakness and muscle wasting (a decrease in muscle strength and size) caused by degenerating muscle fibers begins in the upper legs and pelvis before spreading into the upper arms. Other symptoms include loss of some reflexes, a waddling gait, frequent falls and clumsiness (especially when running), difficulty when rising from a sitting or lying position or when climbing stairs, changes to overall posture, impaired breathing, lung weakness, and cardiomyopathy (heart muscle weakness that interferes with pumping ability). Many children are unable to run or jump. The wasting muscles, in particular the calf muscle (and, less commonly, muscles in the buttocks, shoulders, and arms), may be enlarged by an accumulation of fat and connective tissue, causing them to look larger and healthier than they actually are (called pseudohypertrophy). As the disease progresses, the muscles in the diaphragm that assist in breathing and coughing may weaken. Patients may experience breathing difficulties, respiratory infections, and swallowing problems. Bone thinning and scoliosis (curving of the spine) are common. Some children are mildly mentally impaired. Between ages 3 and 6, children may show brief periods of physical improvement followed by progressive muscle degeneration. Children with Duchenne MD are typically wheelchair-bound by age 12 and usually die in their late teens or early twenties from progressive weakness of the heart muscle, respiratory complications, or infection. Duchenne MD results from an absence of the muscle protein dystrophin. And blood tests of children with Duchenne MD show an abnormally high level of creatine kinase, which is apparent from birth. A rare, autosomal recessive form of MD is seen primarily in the Middle East and North Africa. The disease is clinically similar to Duchenne but is less severe and progresses more slowly. Onset of muscle weakness is typically between ages 5 and 10. Most patients lose the ability to walk in their early twenties, and most die in their forties from cardiac or respiratory complications. Becker MD is less severe than but closely related to Duchenne MD. Persons with Becker MD have partial but insufficient function of the protein dystrophin. The disorder usually appears around age 11 but may occur as late as age 25, and patients generally live into middle age or later. The rate of progressive, symmetric (on both sides of the body) muscle atrophy and weakness varies greatly among affected individuals. Many patients are able to walk until they are in their mid-thirties or later, while others are unable to walk past their teens. Some affected individuals never need to use a wheelchair. As in Duchenne MD, muscle weakness in Becker MD is typically noticed first in the upper arms and shoulders, upper legs, and pelvis. Early symptoms of Becker MD include walking on one's toes, frequent falls, and difficulty rising from the floor. Calf muscles may appear large and healthy as deteriorating muscle fibers are replaced by fat, and muscle activity may cause cramps in some people. Cardiac and mental impairments are not as severe as in Duchenne MD. Congenital MD refers to a group of autosomal recessive muscular dystrophies that are either present at birth or become evident before age 2. They affect both boys and girls. The degree and progression of muscle weakness and degeneration vary with the type of disorder. Weakness may be first noted when children fail to meet landmarks in motor function and muscle control. Muscle degeneration may be mild or severe and is restricted primarily to skeletal muscle. The majority of patients are unable to sit or stand without support, and some affected children may never learn to walk. There are three groups of congenital MD:
Defects in the protein merosin cause nearly half of all cases of congenital MD. Patients with congenital MD may develop contractures (chronic shortening of muscles or tendons around joints, which prevents the joints from moving freely), scoliosis, respiratory and swallowing difficulties, and foot deformities. Some patients have normal intellectual development while others become severely impaired. Weakness in diaphragm muscles may lead to respiratory failure. Congenital MD may also affect the central nervous system, causing vision and speech problems, seizures, and structural changes in the brain. Some children with the disorders die in infancy while others may live into adulthood with only minimal disability. Emery-Dreifuss MD primarily affects boys. The disorder has two forms: one is X-linked recessive and the other is autosomal dominant. Onset of Emery-Dreifuss MD is usually apparent by age 10, but symptoms can appear as late as the mid-twenties. This disease causes slow but progressive wasting of the upper arm and lower leg muscles and symmetric weakness. Contractures in the spine, ankles, knees, elbows, and back of the neck usually precede significant muscle weakness, which is less severe than in Duchenne MD. Contractures may cause elbows to become locked in a flexed position. The entire spine may become rigid as the disease progresses. Other symptoms include shoulder deterioration, toe-walking, and mild facial weakness. Serum creatine kinase levels may be moderately elevated. Nearly all Emery-Dreifuss MD patients have some form of heart problem by age 30, often requiring a pacemaker or other assistive device. Female carriers of the disorder often have cardiac complications without muscle weakness. Patients often die in mid-adulthood from progressive pulmonary or cardiac failure. Youth/adolescent-onset muscular dystrophies are classified two ways: Facioscapulohumeral MD (FSHD) initially affects muscles of the face (facio), shoulders (scapulo), and upper arms (humera) with progressive weakness. Also known as Landouzy-Dejerine disease, this third most common form of MD is an autosomal dominant disorder. Life expectancy is normal, but some individuals become severely disabled. Disease progression is typically very slow, with intermittent spurts of rapid muscle deterioration. Onset is usually in the teenage years but may occur as late as age 40. Muscles around the eyes and mouth are often affected first, followed by weakness around the lower shoulders and chest. A particular pattern of muscle wasting causes the shoulders to appear to be slanted and the shoulder blades to appear winged. Muscles in the lower extremities may also become weakened. Reflexes are impaired only at the biceps and triceps. Changes in facial appearance may include the development of a crooked smile, a pouting look, flattened facial features, or a mask-like appearance. Some patients cannot pucker their lips or whistle and may have difficulty swallowing, chewing, or speaking. Other symptoms may include hearing loss (particularly at high frequencies) and lordosis, an abnormal swayback curve in the spine. Contractures are rare. Some FSHD patients feel severe pain in the affected limb. Cardiac muscles are not affected, and the pelvic girdle is rarely significantly involved. An infant-onset form of FSHD can also cause retinal disease and some hearing loss. Limb-girdle MD refers to more than a dozen inherited conditions marked by progressive loss of muscle bulk and symmetrical weakening of voluntary muscles, primarily those in the shoulders and around the hips. At least three forms of autosomal dominant limb-girdle MD (known as type 1) and eight forms of autosomal recessive limb-girdle MD (known as type 2) have been identified. Some autosomal recessive forms of the disorder are now known to be due to a deficiency of any of four dystrophin-glycoprotein complex proteins called the sarcoglycans. The recessive limb-girdle muscular dystrophies occur more frequently than the dominant forms, usually begin in childhood or the teenage years, and show dramatically increased levels of serum creatine kinase. The dominant limb-girdle muscular dystrophies usually begin in adulthood. In general, the earlier the clinical signs appear, the more rapid the rate of disease progression. Limb-girdle MD affects both males and females. Some forms of the disease progress rapidly, resulting in serious muscle damage and loss of the ability to walk, while others advance very slowly over many years and cause minimal disability, allowing a normal life expectancy. In some cases, the disorder appears to halt temporarily, but symptoms then resume. Weakness is typically noticed first around the hips before spreading to the shoulders, legs, and neck. Patients develop a waddling gait and have difficulty when rising from chairs, climbing stairs, or carrying heavy objects. Patients fall frequently and are unable to run. Contractures at the elbows and knees are rare but patients may develop contractures in the back muscles, which gives them the appearance of a rigid spine. Proximal reflexes (closest to the center of the body) are often impaired. Some patients also experience cardiomyopathy and respiratory complications. Intelligence remains normal. Most persons with limb-girdle MD become severely disabled within 20 years of disease onset. There are three forms of MD that usually begin in adulthood. Distal MD, also called distal myopathy, describes a group of at least six specific muscle diseases that primarily affect distal muscles (those farthest away from the shoulders and hips) in the forearms, hands, lower legs, and feet. Distal dystrophies are typically less severe, progress more slowly, and involve fewer muscles than other forms of MD, although they can spread to other muscles. Distal MD can affect the heart and respiratory muscles, and patients may eventually require the use of a ventilator. Patients may not be able to perform fine hand movement and have difficulty extending the fingers. As leg muscles become affected, walking and climbing stairs become difficult and some patients may be unable to hop or stand on their heels. Onset of distal MD, which affects both men and women, is typically between the ages of 40 and 60 years. In one form of distal MD, a muscle membrane protein complex called dysferlin is known to be lacking. Although distal MD is primarily an autosomal dominant disorder, autosomal recessive forms have been reported in young adults. Symptoms are similar to those of Duchenne MD but with a different pattern of muscle damage. An infantile-onset form of autosomal recessive distal MD has also been reported. Slow but progressive weakness is often first noticed around age 1, when the child begins to walk, and continues to progress very slowly throughout adult life. Myotonic MD, also known as Steinert's disease and dystrophia myotonica, may be the most common adult form of MD. Myotonia, or an inability to relax muscles following a sudden contraction, is found only in this form of MD. People with myotonic MD can live a long life, with variable but slowly progressive disability. Typical disease onset is between ages 20 and 30, but it may develop earlier. Myotonic MD affects the central nervous system and other body systems, including the heart, adrenal glands and thyroid, eyes, and gastrointestinal tract. Muscles in the face and the front of the neck are usually first to show weakness and may produce a haggard, "hatchet" face and a thin, swan-like neck. Wasting and weakness noticeably affect forearm muscles. Other symptoms include cardiac complications, difficulty swallowing, droopy eyelids (called ptosis), cataracts, poor vision, early frontal baldness, weight loss, impotence, testicular atrophy, mild mental impairment, and increased sweating. Patients may also feel drowsy and have an excess need to sleep. This autosomal dominant disease affects both men and women. Females may have irregular menstrual periods and may be infertile. The disease occurs earlier and is more severe in successive generations. A childhood form of myotonic MD may become apparent between ages 5 and 10. Symptoms include general muscle weakness (particularly in the face and distal muscles), lack of muscle tone, and mental impairment. An expectant mother with myotonic MD can give birth to an infant with a rare congenital form of the disorder. Symptoms at birth may include difficulty swallowing or sucking, impaired breathing, absence of reflexes, skeletal deformities (such as club feet), and noticeable muscle weakness, especially in the face. Children with congenital myotonic MD may also experience mental impairment and delayed motor development. This severe infantile form of myotonic MD occurs almost exclusively in children who have inherited the defective gene from their mother, who may not know she is a carrier of the disease. The inherited gene defect that causes myotonic MD is an abnormally long repetition of a three-letter "word" in the genetic code. In unaffected people, the word is repeated a number of times, but in people with myotonic MD, it is repeated many more times. This triplet repeat gets longer with each successive generation. The triplet repeat mechanism has now been implicated in at least 15 other disorders, including Huntington's disease and the spinocerebellar ataxias. Oculopharyngeal MD (OPMD) generally begins in a person's forties or fifties and affects both men and women. In the United States, the disease is most common in families of French-Canadian descent and among Hispanic residents of northern New Mexico. Patients first report drooping eyelids, followed by weakness in the facial muscles and pharyngeal muscles in the throat, causing difficulty swallowing. The tongue may atrophy and changes to the voice may occur. Eyelids may droop so dramatically that some patients compensate by tilting back their heads. Patients may have double vision and problems with upper gaze, and others may have retinitis pigmentosa (progressive degeneration of the retina that affects night vision and peripheral vision) and cardiac irregularities. Muscle weakness and wasting in the neck and shoulder region is common. Limb muscles may also be affected. Persons with OPMD may find it difficult to walk, climb stairs, kneel, or bend. Those persons most severely affected will eventually lose the ability to walk. How are the muscular dystrophies diagnosed?Both the patient's medical history and a complete family history should be thoroughly reviewed to determine if the muscle disease is secondary to a disease affecting other tissues or organs or is an inherited condition. It is also important to rule out any muscle weakness resulting from prior surgery, exposure to toxins, or current medications that may affect the patient's functional status. Thorough clinical and neurological exams can rule out disorders of the central and/or peripheral nervous systems, identify any patterns of muscle weakness and atrophy, test reflex responses and coordination, and look for contractions. Various laboratory tests may be used to confirm the diagnosis of MD. Blood and urine tests can detect defective genes and help identify specific neuromuscular disorders. For example:
Electron microscopy can identify changes in subcellular components of muscle fibers. Electron microscopy can also identify changes that characterize cell death, mutations in muscle cell mitochondria, and an increase in connective tissue seen in muscle diseases such as MD. Changes in muscle fibers that are evident in a rare form of distal MD can be seen using an electron microscope. Exercise tests can detect elevated rates of certain chemicals following exercise and are used to determine the nature of the MD or other muscle disorder. Some exercise tests can be performed at the patient's bedside while others are done at clinics or other sites using sophisticated equipment. These tests also assess muscle strength. They are performed when the patient is relaxed and in the proper position to allow technicians to measure muscle function against gravity and detect even slight muscle weakness. If weakness in respiratory muscles is suspected, respiratory capacity may be measured by having the patient take a deep breath and count slowly while exhaling. Genetic testing looks for genes known to either cause or be associated with inherited muscle disease. DNA analysis and enzyme assays can confirm the diagnosis of certain neuromuscular diseases, including MD. Genetic linkage studies can identify whether a specific genetic marker on a chromosome and a disease are inherited together. They are particularly useful in studying families with members in different generations who are affected. An exact molecular diagnosis is necessary for some of the treatment strategies that are currently being developed. Genetic counseling can help parents who have a family history of MD determine if they are carrying one of the mutated genes that cause the disorder. Two tests can be used to help expectant parents find out if their child is affected.
Magnetic resonance imaging (MRI) is used to examine muscle quality, any atrophy or abnormalities in size, and fatty replacement of muscle tissue, as well as to monitor disease progression. MRI scanning equipment creates a strong magnetic field around the body. Radio waves are then passed through the body to trigger a resonance signal that can be detected at different angles within the body. A computer processes this resonance into either a three-dimensional picture or a two-dimensional "slice" of the tissue being scanned. MRI is a noninvasive, painless procedure. Other forms of diagnostic imaging for MD include phosphorus magnetic resonance spectroscopy, which measures cellular response to exercise and the amount of energy available to muscle fiber, and ultrasound imaging (also known as sonography), which uses high-frequency sound waves to obtain images inside the body. The sound wave echoes are recorded and displayed on a computer screen as a real-time visual image. Ultrasound may be used to measure muscle bulk. Muscle biopsies are used to monitor the course of disease and treatment effectiveness. Using a local anesthetic, a small sample of muscle is removed and studied under a microscope. The sample may be gathered either surgically, through a slit made in the skin, or by needle biopsy, in which a thin hollow needle is inserted through the skin and into the muscle. A small piece of muscle remains in the hollow needle when it is removed from the body. The muscle specimen is stained and examined to determine whether the patient has muscle disease, nerve disease (neuropathy), inflammation, or another myopathy. Muscle biopsies also assist in carrier testing. With the advent of accurate molecular techniques, muscle biopsy is no longer essential for diagnosis. Immunofluorescence testing can detect specific proteins such as dystrophin within muscle fibers. Following biopsy, fluorescent markers are used to stain the sample that has the protein of interest. Neurophysiology studies can identify physical and/or chemical changes in the nervous system.
How are the muscular dystrophies treated?There is no specific treatment that can stop or reverse the progression of any form of MD. All forms of MD are genetic and cannot be prevented. Treatment is aimed at keeping the patient independent for as long as possible and preventing complications that result from weakness, reduced mobility, and cardiac and respiratory difficulties. Treatment may involve a combination of approaches, including physical therapy, drug therapy, and surgery. Assisted ventilation is often needed to treat respiratory muscle weakness that accompanies many forms of MD, especially in the later stages. Oxygen is fed through a flexible mask (or, in some cases, a tube inserted through the esophagus and into the lungs) to help the lungs inflate fully. Since respiratory difficulty may be most extreme at night, some patients may need overnight ventilation. A mask worn over the face is connected by tube to a machine that puts out intermittent bursts of forced oxygen. Obese patients with Duchenne MD may develop obstructive sleep apnea and require nighttime ventilation. Patients on a ventilator may also require the use of a gastric feeding tube. Drug therapy may be prescribed to delay muscle degeneration. Corticosteroids such as prednisone can slow the rate of muscle deterioration in Duchenne MD and help children retain strength and prolong independent walking by as much as several years. However, these medicines have side effects such as weight gain and bone fragility that can be especially troubling in children. Immunosuppressive drugs such as cyclosporin and azathioprine can delay some damage to dying muscle cells. Drugs that may provide short-term relief from myotonia (muscle spasms and weakness) include mexiletine; phenytoin; baclofen, which blocks signals sent from the spinal cord to contract the muscles; dantrolene, which interferes with the process of muscle contraction; and quinine. (Drugs for myotonia are not effective in myotonic MD but work well for myotonia congenita, a genetic neuromuscular disorder characterized by the slow relaxation of the muscles.) Anticonvulsants, also known as antiepileptics, are used to control seizures and some muscle activity. Commonly prescribed oral anticonvulsants include carbamazepine, phenytoin, clonazepam, gabapentin, topiramate, and felbamate. Respiratory infections may be treated with antibiotics. Physical therapy can help prevent deformities, improve movement, and keep muscles as flexible and strong as possible. Options include passive stretching, postural correction, and exercise. A program is developed to meet the individual patient's needs. Therapy should begin as soon as possible following diagnosis, before there is joint or muscle tightness.
Dietary changes have not been shown to slow the progression of MD. Proper nutrition is essential, however, for overall health. Limited mobility or inactivity resulting from muscle weakness can contribute to obesity, dehydration, and constipation. A high-fiber, high-protein, low-calorie diet combined with recommended fluid intake may help. MD patients with swallowing or breathing disorders and those persons who have lost the ability to walk independently should be monitored for signs of malnutrition. Occupational therapy may help some patients deal with progressive weakness and loss of mobility. Some individuals may need to learn new job skills or new ways to perform tasks while other persons may need to change jobs. Assistive technology may include modifications to home and workplace settings and the use of motorized wheelchairs, wheelchair accessories, and adaptive utensils. Corrective surgery is often performed to ease complications from MD.
What is the prognosis?The prognosis varies according to the type of MD and the speed of progression. Some types are mild and progress very slowly, allowing normal life expectancy, while others are more severe and result in functional disability and loss of ambulation. Life expectancy may depend on the degree of muscle weakness and any respiratory and/or cardiac complications. What research is being done?The National Institute of Neurological Disorders and Stroke (NINDS) supports a broad program of research on MD. The goals of these studies are to increase understanding of MD and its causes, develop better therapies, and, ultimately, find ways to prevent and cure it. The NINDS and its sister institutes, the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and the National Institute of Child Health and Human Development (NICHD), lead the MD research efforts conducted at the National Institutes of Health (NIH) or at grantee institutions throughout the country. Drug-based
therapy to delay muscle wasting Corticosteroids are known to extend the ability of Duchenne MD patients to walk by up to 2 years, but steroids have substantial side effects and their mechanism of action is unknown. NIH-funded scientists have established clinical standards for steroid treatment of Duchenne MD. A recent study identified one mechanism of steroid action, raising the prospect that a modified steroid may be designed to minimize or eliminate side effects. Researchers at the NINDS and several universities are exploring the potential of using agents that inhibit enzymes that degrade muscle as a treatment for various types of MD. Scientists are also exploring several other drugs that may help delay the loss of muscle mass:
Enhancing
natural muscle repair mechanisms NIH-supported studies have provided a broad understanding of the mechanisms of muscle regeneration. Additional NIH-funded studies are identifying points in the regeneration-repair pathways that can be targeted by either drug or gene therapy for muscle rescue. For example, researchers have shown that chronic blockade of the muscle growth inhibitor myostatin can enhance muscle repair in animal models of MD. Other NIH-funded investigators have found that increased expression of a muscle repair protein, dysferlin, can help offset muscle damage in dystrophic animals. And the strategy of enhancing natural muscle repair mechanisms with insulin-like growth factor 2 is being used in a clinical trial for myotonic dystrophy. If effective, this approach is translatable to other types of MD. Cell-based
therapy The natural regenerative capacity of muscle provides possibilities for treatment of MD. Attempts to take muscle precursor cells from fathers of Duchenne patients and implant them into patients' muscles originally failed. However, more recent studies have focused on using stem cells to try to restore missing proteins in MD patients. Researchers have shown that stem cells can be used to deliver a functional dystrophin gene to skeletal muscles of dystrophic mice. Gene
replacement therapy Over the last several years, a mini-dystrophin gene (one that is small enough for a viral carrier to deliver it) has proven successful in animal models of Duchenne MD. Viral delivery systems are much better today than they once were (viral vector delivery can set off a serious immune response). As a result, NIH-funded researchers have made important progress in delivering a dystrophin mini-gene to muscles of a mouse model of Duchenne MD. Scientists also are using high-throughput screening (HTS) to find drugs that increase the muscle production of the protein utrophin, which is similar to dystrophin and can help compensate for its loss. HTS lets scientists test hundreds of chemical compounds quickly to find leads for further drug development. Genetic
modification therapy to bypass inherited mutations Two strategies are currently under study to bypass dystrophin mutations. First, the antibiotic gentamicin has been shown to be effective in causing the synthesis machinery to ignore the premature stop signal and produce functional dystrophin. This strategy may be useful in about 15 percent of Duchenne MD patients. An NINDS-funded clinical trial using gentamicin in Duchenne MD patients is under way. Second, a more recent approach uses splicing technology to skip past the mutations in the dystrophin gene to a point where the genetic information is complete and can produce a functional protein. This strategy has shown promise in a mouse model of Duchenne MD. As many as 80 percent of patients could benefit from this new technology. Moving
forward with research in MD The NINDS and the NIAMS fund a research registry for FSHD and myotonic MD. This national registry serves as a resource for scientists seeking a cure for these diseases, in addition to enhancing research on what changes occur in MD. The registry, based at the University of Rochester in New York, recruits patients and stores medical and family history data for individuals with clinically diagnosed FSHD and myotonic MD. Scientists have access to statistical analyses of the registry data and to registry members who have agreed to assist with particular research studies. Similar registries for Duchenne MD are supported by the Centers for Disease Control and Prevention. The
MD CARE Act and the federal commitment to muscular dystrophy In response to the MD CARE Act, the NIH formed the Muscular Dystrophy Coordinating Committee to help guide research on MD. The MD Coordinating Committee is made up of physicians, scientists, NIH professional staff, and representatives of other federal agencies and voluntary health organizations with a focus on MD. The purpose of the group is to help NIH add new capabilities to the national effort to understand and treat MD, without duplicating existing programs. The MD Coordinating Committee has developed a broad research and education plan and continues to refine the plan to improve basic, translational, and clinical research in MD, with the goal of improving the quality of life for patients with MD. Information about the committee and plan is available at http://www.ninds.nih.gov/find_people/groups/mdcc/. Past research has led to the discovery of disease mechanisms and improved treatment for many forms of MD. Current research promises to generate further improvements. In the coming years, physicians and patients can look forward to new forms of therapy developed through an understanding of the unique traits of MD. Where can I get more information? For more information on neurological disorders or research programs
funded by the National Institute of Neurological Disorders and
Stroke, contact the Institute's Brain Resources and Information
Network (BRAIN) at: BRAIN Information also is available from the following organizations:
amniocentesis - a process of prenatal genetic analysis that uses a sample of the amniotic fluid taken from the womb. atrophy - a decrease in size or wasting away of a body part or tissue. autosomal dominant - a pattern of inheritance in which a child acquires a disease by receiving a normal gene from one parent and a defective gene from the other parent. autosomal recessive - a pattern of inheritance in which both parents carry and pass on a defective gene to their child. biopsy - a procedure in which tissue or other material is removed from the body and studied for signs of disease. cardiomyopathy - heart muscle weakness that interferes with the heart's ability to pump blood. carrier - an individual who doesn't have a disease but has one normal gene and one gene for a genetic disorder and is therefore capable of passing this disease to her or his children. chorionic villus sampling - a prenatal genetic test that involves removal and examination of a piece of the placenta. chromosomes - genetic structures that contains DNA. contracture - chronic shortening of a muscle or tendon that limits movement of a bony joint, such as the elbow. creatine kinase - a protein needed for the chemical reactions that produce energy for muscle contractions; high levels in the blood indicate muscle damage. dystrophin - a protein that helps maintain the shape and structure of muscle fibers. electromyography - a recording and study of the electrical properties of skeletal muscle. glycoprotein - a molecule that has a protein and a carbohydrate component. linkage studies - tests conducted among family members to determine how a genetic trait is passed on through generations. lordosis - an abnormal forward curving of the spine. merosin - a protein found in the connective tissue that surrounds muscle fibers. myoglobin - an oxygen-binding protein in muscle cells that generates energy by turning glucose into carbon dioxide and water. myopathy - any disorder of muscle tissue or muscles. myotonia - an inability to relax muscles following a sudden contraction. phagocytosis - a process in which material is taken into the cell and digested. pseudohypertrophy -a condition in which muscles may be enlarged by an accumulation of fat and connective tissue, causing them to look larger and healthier than they actually are. ptosis - an abnormal drooping of the eyelids. scoliosis - an abnormal lateral, or sideways, curving of the spine. X-linked recessive - a pattern of disease inheritance in which the mother carries the affected gene on the chromosome that determines the child's sex and passes it to her son.
|
|
Neuropathy- any of numerous disturbances or pathologic changes in the peripheral nervous system; see also mononeuropathy and polyneuropathy. The etiology may be known (such as arsenic, diabetic, ischemic, or traumatic neuropathy) or unknown. Encephalopathy and myelopathy are corresponding terms relating to involvement of the brain and spinal cord. The term is also used to designate noninflammatory lesions in the peripheral nervous system, in contrast to inflammatory lesions (neuritis). adj., neuropath´ic., adj. (Dorland's Medical Dictionary) Diabetic
Neuropathy
|
|||||||||||||||||||||||||||||
|
What
is Diabetic Neuropathy?
Is there any treatment? What is the prognosis? What research is being done? Clinical Trials Organizations Additional resources from MEDLINEplus What is Diabetic Neuropathy?
Diabetic neuropathy is a peripheral nerve disorder caused by diabetes or poor blood sugar control. The most common types of diabetic neuropathy result in problems with sensation in the feet. It can develop slowly after many years of diabetes or may occur early in the disease. The symptoms are numbness, pain, or tingling in the feet or lower legs. The pain can be intense and require treatment to relieve the discomfort. The loss of sensation in the feet may also increase the possibility that foot injuries will go unnoticed and develop into ulcers or lesions that become infected. In some cases, diabetic neuropathy can be associated with difficulty walking and some weakness in the foot muscles. There are other types of diabetic-related neuropathies that affect specific parts of the body. For example, diabetic amyotrophy causes pain, weakness and wasting of the thigh muscles, or cranial nerve infarcts that may result in double vision, a drooping eyelid, or dizziness. Diabetes can also affect the autonomic nerves that control blood pressure, the digestive tract, bladder function, and sexual organs. Problems with the autonomic nerves may cause lightheadedness, indigestion, diarrhea or constipation, difficulty with bladder control, and impotence. Is there any treatment?
The goal of treating diabetic neuropathy is to prevent further tissue damage and relieve discomfort. The first step is to bring blood sugar levels under control by diet and medication. Another important part of treatment involves taking special care of the feet by wearing proper fitting shoes and routinely checking the feet for cuts and infections. Analgesics, low doses of antidepressants, and some anticonvulsant medications may be prescribed for relief of pain, burning, or tingling. Some individuals find that walking regularly, taking warm baths, or using elastic stockings may help relieve leg pain. What is the prognosis?
The prognosis for diabetic neuropathy depends largely on how well the underlying condition of diabetes is handled. Treating diabetes may halt progression and improve symptoms of the neuropathy, but recovery is slow. The painful sensations of diabetic neuropathy may become severe enough to cause depression in some patients. What research is being done?
The NINDS conducts and supports research on diabetic neuropathy to increase understanding of the disorder and find ways to prevent and cure it. New medications are currently being examined to assess improvement or stabilization of neuropathic symptoms. NIH Patient Recruitment for Diabetic Neuropathy Clinical Trials
What
are Hereditary Neuropathies?
Is there any treatment? What is the prognosis? What research is being done? Clinical Trials Organizations Related NINDS Publications and Information What are Hereditary Neuropathies?
Hereditary neuropathies are a group of inherited disorders affecting the peripheral nervous system. The hereditary neuropathies are divided into four major subcategories: hereditary motor and sensory neuropathy, hereditary sensory neuropathy, hereditary motor neuropathy, and hereditary sensory and autonomic neuropathy. The most common type is Charcot-Marie-Tooth disease, one of the hereditary motor and sensory neuropathies. Symptoms of the hereditary neuropathies vary according to the type and may include sensory symptoms such as numbness, tingling, and pain in the feet and hands; or motor symptoms such as weakness and loss of muscle bulk, particularly in the lower leg and feet muscles. Certain types of hereditary neuropathies can affect the autonomic nerves, resulting in impaired sweating, postural hypotension, or insensitivity to pain. Some people may have foot deformities such as high arches and hammer toes, thin calf muscles (having the appearance of an inverted champagne glass) or scoliosis (curvature of the spine). The symptoms of hereditary neuropathies may be apparent at birth or appear in middle or late life. They can vary among different family members, with some family members being more severely affected than others. The hereditary neuropathies can be diagnosed by blood tests for genetic testing, nerve conduction studies, and nerve biopsies. Is there any treatment?
There are no standard treatments for hereditary neuropathies. Treatment is mainly symptomatic and supportive. Medical treatment includes physical therapy and if needed, pain medication. Orthopedic surgery may be needed to correct severe foot or other skeletal deformities. Bracing may also be used to improve mobility. What is the prognosis?
The prognosis for individuals with hereditary neuropathies depends upon the type of neuropathy. Some hereditary neuropathies have very mild symptoms and may go undiagnosed for many years. Other types are more severe and are associated with more disabilities. Genetic counseling is important to understand further details about the disease and prognosis. What research is being done?
The NINDS supports research on neuromuscular disorders, such as hereditary neuropathies, aimed at learning more about these disorders and finding ways to prevent and treat them. NIH Patient Recruitment for Hereditary Neuropathies Clinical Trials
Related
NINDS Publications and Information
|
|
What
is Charcot-Marie-Tooth disease?
What are the symptoms of Charcot-Marie-Tooth disease? What are the types of Charcot-Marie-Tooth disease? What causes Charcot-Marie-Tooth disease? How is Charcot-Marie-Tooth disease diagnosed? How is Charcot-Marie-Tooth disease treated? What research is being done? Where can I get more information? What is Charcot-Marie-Tooth disease?Charcot-Marie-Tooth disease (CMT) is one of the most common inherited neurological disorders, affecting approximately 1 in 2,500 people in the United States. The disease is named for the three physicians who first identified it in 1886 - Jean-Martin Charcot and Pierre Marie in Paris, France, and Howard Henry Tooth in Cambridge, England. CMT, also known as hereditary motor and sensory neuropathy (HMSN) or peroneal muscular atrophy, comprises a group of disorders that affect peripheral nerves. The peripheral nerves lie outside the brain and spinal cord and supply the muscles and sensory organs in the limbs. Disorders that affect the peripheral nerves are called peripheral neuropathies. What are the symptoms of Charcot-Marie-Tooth disease?The neuropathy of CMT affects both motor and sensory nerves. A typical feature includes weakness of the foot and lower leg muscles, which may result in foot drop and a high-stepped gait with frequent tripping or falls. Foot deformities, such as high arches and hammertoes (a condition in which the middle joint of a toe bends upwards) are also characteristic due to weakness of the small muscles in the feet. In addition, the lower legs may take on an "inverted champagne bottle" appearance due to the loss of muscle bulk. Later in the disease, weakness and muscle atrophy may occur in the hands, resulting in difficulty with fine motor skills. Onset of symptoms is most often in adolescence or early adulthood, however presentation may be delayed until mid-adulthood. The severity of symptoms is quite variable in different patients and even among family members with the disease. Progression of symptoms is gradual. Pain can range from mild to severe, and some patients may need to rely on foot or leg braces or other orthopedic devices to maintain mobility. Although in rare cases patients may have respiratory muscle weakness, CMT is not considered a fatal disease and people with most forms of CMT have a normal life expectancy. What are the types of Charcot-Marie-Tooth disease?There are many forms of CMT disease, including CMT1, CMT2, CMT3, CMT4, and CMTX. CMT1, caused by abnormalities in the myelin sheath, has three main types. CMT1A is an autosomal dominant disease resulting from a duplication of the gene on chromosome 17 that carries the instructions for producing the peripheral myelin protein-22 (PMP-22). The PMP-22 protein is a critical component of the myelin sheath. An overabundance of this gene causes the structure and function of the myelin sheath to be abnormal. Patients experience weakness and atrophy of the muscles of the lower legs beginning in adolescence; later they experience hand weakness and sensory loss. Interestingly, a different neuropathy distinct from CMT1A called hereditary neuropathy with predisposition to pressure palsy (HNPP) is caused by a deletion of one of the PMP-22 genes. In this case, abnormally low levels of the PMP-22 gene result in episodic, recurrent demyelinating neuropathy. CMT1B is an autosomal dominant disease caused by mutations in the gene that carries the instructions for manufacturing the myelin protein zero (P0), which is another critical component of the myelin sheath. Most of these mutations are point mutations, meaning a mistake occurs in only one letter of the DNA genetic code. To date, scientists have identified more than 30 different point mutations in the P0 gene. As a result of abnormalities in P0, CMT1B produces symptoms similar to those found in CMT1A. The gene defect that causes CMT1C, which also has symptoms similar to those found in CMT1A, has not yet been identified. CMT2 results from abnormalities in the axon of the peripheral nerve cell rather than the myelin sheath. There are many subtypes of CMT2, designated by the letters from A-L. Each subtype is characterized by the mode of inheritance and associated clinical features. The genetic loci have been identified for some subtypes. Recently, a mutation was identified in the gene that codes for the kinesin family member 1B-beta protein in families with CMT2A. Kinesins are proteins that act as motors to help power the transport of materials along the train tracks (microtubules) of the cell. Another recent finding is a mutation in the neurofilament-light gene, identified in a Russian family with CMT2E. Neurofilaments are structural proteins that help maintain the normal shape of a cell. CMT3 or Dejerine-Sottas disease is a severe demyelinating neuropathy that begins in infancy. Infants have severe muscle atrophy, weakness, and sensory problems. This rare disorder can be caused by a specific point mutation in the P0 gene or a point mutation in the PMP-22 gene. CMT4 comprises several different subtypes of autosomal recessive demyelinating motor and sensory neuropathies. Each neuropathy subtype is caused by a different genetic mutation, may affect a particular ethnic population, and produces distinct physiologic or clinical characteristics. Patients with CMT4 generally develop symptoms of leg weakness in childhood and by adolescence they may not be able to walk. The gene abnormalities responsible for CMT4 have yet to be identified. CMTX is an X-linked dominant disease and is caused by a point mutation in the connexin-32 gene on the X chromosome. The connexin-32 protein is expressed in Schwann cells-cells that wrap around nerve axons, making up a single segment of the myelin sheath. This protein may be involved in Schwann cell communication with the axon. Males who inherit one mutated gene from their mothers show moderate to severe symptoms of the disease beginning in late childhood or adolescence (the Y chromosome that males inherit from their fathers does not have the connexin-32 gene). Females who inherit one mutated gene from one parent and one normal gene from the other parent may develop mild symptoms in adolescence or later or may not develop symptoms of the disease at all. What causes Charcot-Marie-Tooth disease?A nerve cell communicates information to distant targets by sending electrical signals down a long, thin part of the cell called the axon. In order to increase the speed at which these electrical signals travel, the axon is insulated by myelin, which is produced by another type of cell called the Schwann cell. Myelin twists around the axon like a jelly-roll cake and prevents dissipation of the electrical signals. Without an intact axon and myelin sheath, peripheral nerve cells are unable to activate target muscles or relay sensory information from the limbs back to the brain. CMT is caused by mutations in genes that produce proteins involved in the structure and function of either the peripheral nerve axon or the myelin sheath. Although different proteins are abnormal in different forms of CMT disease, all of the mutations affect the normal function of the peripheral nerves. Consequently, these nerves slowly degenerate and lose the ability to communicate with their distant targets. The degeneration of motor nerves results in muscle weakness and atrophy in the extremities (arms, legs, hands, or feet), and in some cases the degeneration of sensory nerves results in a reduced ability to feel heat, cold, and pain. The gene mutations in CMT disease are usually inherited. Each of us normally possesses two copies of every gene, one inherited from each parent. Some forms of CMT are inherited in an autosomal dominant fashion, which means that only one copy of the abnormal gene is needed to cause the disease. Other forms of CMT are inherited in an autosomal recessive fashion, which means that both copies of the abnormal gene must be present to cause the disease. Still other forms of CMT are inherited in an X-linked fashion, which means that the abnormal gene is located on the X chromosome. The X and Y chromosomes determine an individual's sex. Individuals with two X chromosomes are female and individuals with one X and one Y chromosome are male. In rare cases the gene mutation causing CMT disease is a new mutation which occurs spontaneously in the patient's genetic material and has not been passed down through the family. How is Charcot-Marie-Tooth disease diagnosed?Diagnosis of CMT begins with a standard patient history, family history, and neurological examination. Patients will be asked about the nature and duration of their symptoms and whether other family members have the disease. During the neurological examination a physician will look for evidence of muscle weakness in the arms, legs, hands, and feet, decreased muscle bulk, reduced tendon reflexes, and sensory loss. Doctors look for evidence of foot deformities, such as high arches, hammertoes, inverted heel, or flat feet. Other orthopedic problems, such as mild scoliosis or hip dysplasia, may also be present. A specific sign that may be found in patients with CMT1 is nerve enlargement that may be felt or even seen through the skin. These enlarged nerves, called hypertrophic nerves, are caused by abnormally thickened myelin sheaths. If CMT is suspected, the physician may order electrodiagnostic tests for the patient. This testing consists of two parts: nerve conduction studies and electromyography (EMG). During nerve conduction studies, electrodes are placed on the skin over a peripheral motor or sensory nerve. These electrodes produce a small electric shock that may cause mild discomfort. This electrical impulse stimulates sensory and motor nerves and provides quantifiable information that the doctor can use to arrive at a diagnosis. EMG involves inserting a needle electrode through the skin to measure the bioelectrical activity of muscles. Specific abnormalities in the readings signify axon degeneration. EMG may be useful in further characterizing the distribution and severity of peripheral nerve involvement. If all other tests seem to suggest that a patient has CMT, a neurologist may perform a nerve biopsy to confirm the diagnosis. A nerve biopsy involves removing a small piece of peripheral nerve through an incision in the skin. This is most often done by removing a piece of the nerve that runs down the calf of the leg. The nerve is then examined under a microscope. Patients with CMT1 typically show signs of abnormal myelination. Specifically, "onion bulb" formations may be seen which represent axons surrounded by layers of demyelinating and remyelinating Schwann cells. Patients with CMT2 usually show signs of axon degeneration. Genetic testing is available for some types of CMT and may soon be available for other types; such testing can be used to confirm a diagnosis. In addition, genetic counseling is available to parents who fear that they may pass mutant genes to their children. How is Charcot-Marie-Tooth disease treated?There is no cure for CMT, but physical therapy, occupational therapy, braces and other orthopedic devices, and even orthopedic surgery can help patients cope with the disabling symptoms of the disease. In addition, pain-killing drugs can be prescribed for patients who have severe pain. Physical and occupational therapy, the preferred treatment for CMT, involves muscle strength training, muscle and ligament stretching, stamina training, and moderate aerobic exercise. Most therapists recommend a specialized treatment program designed with the approval of the patient's physician to fit individual abilities and needs. Therapists also suggest entering into a treatment program early; muscle strengthening may delay or reduce muscle atrophy, so strength training is most useful if it begins before nerve degeneration and muscle weakness progress to the point of disability. Stretching may prevent or reduce joint deformities that result from uneven muscle pull on bones. Exercises to help build stamina or increase endurance will help prevent the fatigue that results from performing everyday activities that require strength and mobility. Moderate aerobic activity can help to maintain cardiovascular fitness and overall health. Most therapists recommend low-impact or no-impact exercises, such as biking or swimming, rather than activities such as walking or jogging, which may put stress on fragile muscles and joints. Many CMT patients require ankle braces and other orthopedic devices to maintain everyday mobility and prevent injury. Ankle braces can help prevent ankle sprains by providing support and stability during activities such as walking or climbing stairs. High-top shoes or boots can also give the patient support for weak ankles. Thumb splints can help with hand weakness and loss of fine motor skills. Assistive devices should be used before disability sets in because the devices may prevent muscle strain and reduce muscle weakening. Some CMT patients may decide to have orthopedic surgery to reverse foot and joint deformities. What research is being done?The NINDS supports research on CMT and other peripheral neuropathies in an effort to learn how to better treat, prevent, and even cure these disorders. Ongoing research includes efforts to identify more of the mutant genes and proteins that cause the various disease subtypes, efforts to discover the mechanisms of nerve degeneration and muscle atrophy with the hope of developing interventions to stop or slow down these debilitating processes, and efforts to find therapies to reverse nerve degeneration and muscle atrophy. One promising area of research involves gene therapy experiments. Research with cell cultures and animal models has shown that it is possible to deliver genes to Schwann cells and muscle. Another area of research involves the use of trophic factors or nerve growth factors, such as the hormone androgen, to prevent nerve degeneration. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 07-4897
|
||||||
|
What
is peripheral neuropathy?
How are the peripheral neuropathies classified? What are the symptoms of peripheral nerve damage? What causes peripheral neuropathy? How is peripheral neuropathy diagnosed? What treatments are available? What research is being done? Where can I get more information? What is peripheral neuropathy?Peripheral neuropathy describes damage to the peripheral nervous system, the vast communications network that transmits information from the brain and spinal cord (the central nervous system) to every other part of the body. Peripheral nerves also send sensory information back to the brain and spinal cord, such as a message that the feet are cold or a finger is burned. Damage to the peripheral nervous system interferes with these vital connections. Like static on a telephone line, peripheral neuropathy distorts and sometimes interrupts messages between the brain and the rest of the body. Because every peripheral nerve has a highly specialized function in a specific part of the body, a wide array of symptoms can occur when nerves are damaged. Some people may experience temporary numbness, tingling, and pricking sensations (paresthesia), sensitivity to touch, or muscle weakness. Others may suffer more extreme symptoms, including burning pain (especially at night), muscle wasting, paralysis, or organ or gland dysfunction. People may become unable to digest food easily, maintain safe levels of blood pressure, sweat normally, or experience normal sexual function. In the most extreme cases, breathing may become difficult or organ failure may occur. Some forms of neuropathy involve damage to only one nerve and are called mononeuropathies. More often though, multiple nerves affecting all limbs are affected-called polyneuropathy. Occasionally, two or more isolated nerves in separate areas of the body are affected-called mononeuritis multiplex. In acute neuropathies, such as Guillain-Barré syndrome, symptoms appear suddenly, progress rapidly, and resolve slowly as damaged nerves heal. In chronic forms, symptoms begin subtly and progress slowly. Some people may have periods of relief followed by relapse. Others may reach a plateau stage where symptoms stay the same for many months or years. Some chronic neuropathies worsen over time, but very few forms prove fatal unless complicated by other diseases. Occasionally the neuropathy is a symptom of another disorder. In the most common forms of polyneuropathy, the nerve fibers (individual cells that make up the nerve) most distant from the brain and the spinal cord malfunction first. Pain and other symptoms often appear symmetrically, for example, in both feet followed by a gradual progression up both legs. Next, the fingers, hands, and arms may become affected, and symptoms can progress into the central part of the body. Many people with diabetic neuropathy experience this pattern of ascending nerve damage. How are the peripheral neuropathies classified?More than 100 types of peripheral neuropathy have been identified, each with its own characteristic set of symptoms, pattern of development, and prognosis. Impaired function and symptoms depend on the type of nerves-motor, sensory, or autonomic-that are damaged. Motor nerves control movements of all muscles under conscious control, such as those used for walking, grasping things, or talking. Sensory nerves transmit information about sensory experiences, such as the feeling of a light touch or the pain resulting from a cut. Autonomic nerves regulate biological activities that people do not control consciously, such as breathing, digesting food, and heart and gland functions. Although some neuropathies may affect all three types of nerves, others primarily affect one or two types. Therefore, doctors may use terms such as predominantly motor neuropathy, predominantly sensory neuropathy, sensory-motor neuropathy, or autonomic neuropathy to describe a patient's condition. What are the symptoms of peripheral nerve damage?Symptoms are related to the type of affected nerve and may be seen over a period of days, weeks, or years. Muscle weakness is the most common symptom of motor nerve damage. Other symptoms may include painful cramps and fasciculations (uncontrolled muscle twitching visible under the skin), muscle loss, bone degeneration, and changes in the skin, hair, and nails. These more general degenerative changes also can result from sensory or autonomic nerve fiber loss. Sensory nerve damage causes a more complex range of symptoms because sensory nerves have a wider, more highly specialized range of functions. Larger sensory fibers enclosed in myelin (a fatty protein that coats and insulates many nerves) register vibration, light touch, and position sense. Damage to large sensory fibers lessens the ability to feel vibrations and touch, resulting in a general sense of numbness, especially in the hands and feet. People may feel as if they are wearing gloves and stockings even when they are not. Many patients cannot recognize by touch alone the shapes of small objects or distinguish between different shapes. This damage to sensory fibers may contribute to the loss of reflexes (as can motor nerve damage). Loss of position sense often makes people unable to coordinate complex movements like walking or fastening buttons, or to maintain their balance when their eyes are shut. Neuropathic pain is difficult to control and can seriously affect emotional well-being and overall quality of life. Neuropathic pain is often worse at night, seriously disrupting sleep and adding to the emotional burden of sensory nerve damage. Smaller sensory fibers without myelin sheaths transmit pain and temperature sensations. Damage to these fibers can interfere with the ability to feel pain or changes in temperature. People may fail to sense that they have been injured from a cut or that a wound is becoming infected. Others may not detect pains that warn of impending heart attack or other acute conditions. (Loss of pain sensation is a particularly serious problem for people with diabetes, contributing to the high rate of lower limb amputations among this population.) Pain receptors in the skin can also become oversensitized, so that people may feel severe pain (allodynia) from stimuli that are normally painless (for example, some may experience pain from bed sheets draped lightly over the body). Symptoms of autonomic nerve damage are diverse and depend upon which organs or glands are affected. Autonomic nerve dysfunction can become life threatening and may require emergency medical care in cases when breathing becomes impaired or when the heart begins beating irregularly. Common symptoms of autonomic nerve damage include an inability to sweat normally, which may lead to heat intolerance; a loss of bladder control, which may cause infection or incontinence; and an inability to control muscles that expand or contract blood vessels to maintain safe blood pressure levels. A loss of control over blood pressure can cause dizziness, lightheadedness, or even fainting when a person moves suddenly from a seated to a standing position (a condition known as postural or orthostatic hypotension). Gastrointestinal symptoms frequently accompany autonomic neuropathy. Nerves controlling intestinal muscle contractions often malfunction, leading to diarrhea, constipation, or incontinence. Many people also have problems eating or swallowing if certain autonomic nerves are affected. What causes peripheral neuropathy?Peripheral neuropathy may be either inherited or acquired. Causes of acquired peripheral neuropathy include physical injury (trauma) to a nerve, tumors, toxins, autoimmune responses, nutritional deficiencies, alcoholism, and vascular and metabolic disorders. Acquired peripheral neuropathies are grouped into three broad categories: those caused by systemic disease, those caused by trauma from external agents, and those caused by infections or autoimmune disorders affecting nerve tissue. One example of an acquired peripheral neuropathy is trigeminal neuralgia (also known as tic douloureux), in which damage to the trigeminal nerve (the large nerve of the head and face) causes episodic attacks of excruciating, lightning-like pain on one side of the face. In some cases, the cause is an earlier viral infection, pressure on the nerve from a tumor or swollen blood vessel, or, infrequently, multiple sclerosis. In many cases, however, a specific cause cannot be identified. Doctors usually refer to neuropathies with no known cause as idiopathic neuropathies. Physical injury (trauma) is the most common cause of injury to a nerve. Injury or sudden trauma, such as from automobile accidents, falls, and sports-related activities, can cause nerves to be partially or completely severed, crushed, compressed, or stretched, sometimes so forcefully that they are partially or completely detached from the spinal cord. Less dramatic traumas also can cause serious nerve damage. Broken or dislocated bones can exert damaging pressure on neighboring nerves, and slipped disks between vertebrae can compress nerve fibers where they emerge from the spinal cord. Systemic diseases — disorders that affect the entire body —often cause peripheral neuropathy. These disorders may include: Metabolic and endocrine disorders. Nerve tissues are highly vulnerable to damage from diseases that impair the body's ability to transform nutrients into energy, process waste products, or manufacture the substances that make up living tissue. Diabetes mellitus, characterized by chronically high blood glucose levels, is a leading cause of peripheral neuropathy in the United States. About 60 percent to 70 percent of people with diabetes have mild to severe forms of nervous system damage. Kidney disorders can lead to abnormally high amounts of toxic substances in the blood that can severely damage nerve tissue. A majority of patients who require dialysis because of kidney failure develop polyneuropathy. Some liver diseases also lead to neuropathies as a result of chemical imbalances. Hormonal imbalances can disturb normal metabolic processes and cause neuropathies. For example, an underproduction of thyroid hormones slows metabolism, leading to fluid retention and swollen tissues that can exert pressure on peripheral nerves. Overproduction of growth hormone can lead to acromegaly, a condition characterized by the abnormal enlargement of many parts of the skeleton, including the joints. Nerves running through these affected joints often become entrapped. Vitamin deficiencies and alcoholism can cause widespread damage to nerve tissue. Vitamins E, B1, B6, B12, and niacin are essential to healthy nerve function. Thiamine deficiency, in particular, is common among people with alcoholism because they often also have poor dietary habits. Thiamine deficiency can cause a painful neuropathy of the extremities. Some researchers believe that excessive alcohol consumption may, in itself, contribute directly to nerve damage, a condition referred to as alcoholic neuropathy. Vascular damage and blood diseases can decrease oxygen supply to the peripheral nerves and quickly lead to serious damage to or death of nerve tissues, much as a sudden lack of oxygen to the brain can cause a stroke. Diabetes frequently leads to blood vessel constriction. Various forms of vasculitis (blood vessel inflammation) frequently cause vessel walls to harden, thicken, and develop scar tissue, decreasing their diameter and impeding blood flow. This category of nerve damage, in which isolated nerves in different areas are damaged, is called mononeuropathy multiplex or multifocal mononeuropathy. Connective tissue disorders and chronic inflammation can cause direct and indirect nerve damage. When the multiple layers of protective tissue surrounding nerves become inflamed, the inflammation can spread directly into nerve fibers. Chronic inflammation also leads to the progressive destruction of connective tissue, making nerve fibers more vulnerable to compression injuries and infections. Joints can become inflamed and swollen and entrap nerves, causing pain. Cancers and benign tumors can infiltrate or exert damaging pressure on nerve fibers. Tumors also can arise directly from nerve tissue cells. Widespread polyneuropathy is often associated with the neurofibromatoses, genetic diseases in which multiple benign tumors grow on nerve tissue. Neuromas, benign masses of overgrown nerve tissue that can develop after any penetrating injury that severs nerve fibers, generate very intense pain signals and sometimes engulf neighboring nerves, leading to further damage and even greater pain. Neuroma formation can be one element of a more widespread neuropathic pain condition called complex regional pain syndrome or reflex sympathetic dystrophy syndrome, which can be caused by traumatic injuries or surgical trauma. Paraneoplastic syndromes, a group of rare degenerative disorders that are triggered by a person's immune system response to a cancerous tumor, also can indirectly cause widespread nerve damage. Repetitive stress frequently leads to entrapment neuropathies, a special category of compression injury. Cumulative damage can result from repetitive, forceful, awkward activities that require flexing of any group of joints for prolonged periods. The resulting irritation may cause ligaments, tendons, and muscles to become inflamed and swollen, constricting the narrow passageways through which some nerves pass. These injuries become more frequent during pregnancy, probably because weight gain and fluid retention also constrict nerve passageways. Toxins can also cause peripheral nerve damage. People who are exposed to heavy metals (arsenic, lead, mercury, thallium), industrial drugs, or environmental toxins frequently develop neuropathy. Certain anticancer drugs, anticonvulsants, antiviral agents, and antibiotics have side effects that can include peripheral nerve damage, thus limiting their long-term use. Infections and autoimmune disorders can cause peripheral neuropathy. Viruses and bacteria that can attack nerve tissues include herpes varicella-zoster (shingles), Epstein-Barr virus, cytomegalovirus, and herpes simplex-members of the large family of human herpes viruses. These viruses severely damage sensory nerves, causing attacks of sharp, lightning-like pain. Postherpetic neuralgia often occurs after an attack of shingles and can be particularly painful. The human immunodeficiency virus (HIV), which causes AIDS, also causes extensive damage to the central and peripheral nervous systems. The virus can cause several different forms of neuropathy, each strongly associated with a specific stage of active immunodeficiency disease. A rapidly progressive, painful polyneuropathy affecting the feet and hands is often the first clinically apparent sign of HIV infection. Lyme disease, diphtheria, and leprosy are bacterial diseases characterized by extensive peripheral nerve damage. Diphtheria and leprosy are now rare in the United States, but Lyme disease is on the rise. It can cause a wide range of neuropathic disorders, including a rapidly developing, painful polyneuropathy, often within a few weeks after initial infection by a tick bite. Viral and bacterial infections can also cause indirect nerve damage by provoking conditions referred to as autoimmune disorders, in which specialized cells and antibodies of the immune system attack the body's own tissues. These attacks typically cause destruction of the nerve's myelin sheath or axon (the long fiber that extends out from the main nerve cell body). Some neuropathies are caused by inflammation resulting from immune system activities rather than from direct damage by infectious organisms. Inflammatory neuropathies can develop quickly or slowly, and chronic forms can exhibit a pattern of alternating remission and relapse. Acute inflammatory demyelinating neuropathy, better known as Guillain-Barré syndrome, can damage motor, sensory, and autonomic nerve fibers. Most people recover from this syndrome although severe cases can be life threatening. Chronic inflammatory demyelinating polyneuropathy (CIDP), generally less dangerous, usually damages sensory and motor nerves, leaving autonomic nerves intact. Multifocal motor neuropathy is a form of inflammatory neuropathy that affects motor nerves exclusively; it may be chronic or acute. Inherited forms of peripheral neuropathy are caused by inborn mistakes in the genetic code or by new genetic mutations. Some genetic errors lead to mild neuropathies with symptoms that begin in early adulthood and result in little, if any, significant impairment. More severe hereditary neuropathies often appear in infancy or childhood. The most common inherited neuropathies are a group of disorders collectively referred to as Charcot-Marie-Tooth disease. These neuropathies result from flaws in genes responsible for manufacturing neurons or the myelin sheath. Hallmarks of typical Charcot-Marie-Tooth disease include extreme weakening and wasting of muscles in the lower legs and feet, gait abnormalities, loss of tendon reflexes, and numbness in the lower limbs. How is peripheral neuropathy diagnosed?Diagnosing peripheral neuropathy is often difficult because the symptoms are highly variable. A thorough neurological examination is usually required and involves taking an extensive patient history (including the patient’s symptoms, work environment, social habits, exposure to any toxins, history of alcoholism, risk of HIV or other infectious disease, and family history of neurological disease), performing tests that may identify the cause of the neuropathic disorder, and conducting tests to determine the extent and type of nerve damage. A general physical examination and related tests may reveal the presence of a systemic disease causing nerve damage. Blood tests can detect diabetes, vitamin deficiencies, liver or kidney dysfunction, other metabolic disorders, and signs of abnormal immune system activity. An examination of cerebrospinal fluid that surrounds the brain and spinal cord can reveal abnormal antibodies associated with neuropathy. More specialized tests may reveal other blood or cardiovascular diseases, connective tissue disorders, or malignancies. Tests of muscle strength, as well as evidence of cramps or fasciculations, indicate motor fiber involvement. Evaluation of a patient’s ability to register vibration, light touch, body position, temperature, and pain reveals sensory nerve damage and may indicate whether small or large sensory nerve fibers are affected. Based on the results of the neurological exam, physical exam, patient history, and any previous screening or testing, additional testing may be ordered to help determine the nature and extent of the neuropathy. Computed tomography, or CT scan, is a noninvasive, painless process used to produce rapid, clear two-dimensional images of organs, bones, and tissues. X-rays are passed through the body at various angles and are detected by a computerized scanner. The data is processed and displayed as cross-sectional images, or "slices," of the internal structure of the body or organ. Neurological CT scans can detect bone and vascular irregularities, certain brain tumors and cysts, herniated disks, encephalitis, spinal stenosis (narrowing of the spinal canal), and other disorders. Magnetic resonance imaging (MRI) can examine muscle quality and size, detect any fatty replacement of muscle tissue, and determine whether a nerve fiber has sustained compression damage. The MRI equipment creates a strong magnetic field around the body. Radio waves are then passed through the body to trigger a resonance signal that can be detected at different angles within the body. A computer processes this resonance into either a three-dimensional picture or a two-dimensional "slice" of the scanned area. Electromyography (EMG) involves inserting a fine needle into a muscle to compare the amount of electrical activity present when muscles are at rest and when they contract. EMG tests can help differentiate between muscle and nerve disorders. Nerve conduction velocity (NCV) tests can precisely measure the degree of damage in larger nerve fibers, revealing whether symptoms are being caused by degeneration of the myelin sheath or the axon. During this test, a probe electrically stimulates a nerve fiber, which responds by generating its own electrical impulse. An electrode placed further along the nerve’s pathway measures the speed of impulse transmission along the axon. Slow transmission rates and impulse blockage tend to indicate damage to the myelin sheath, while a reduction in the strength of impulses is a sign of axonal degeneration. Nerve biopsy involves removing and examining a sample of nerve tissue, most often from the lower leg. Although this test can provide valuable information about the degree of nerve damage, it is an invasive procedure that is difficult to perform and may itself cause neuropathic side effects. Many experts do not believe that a biopsy is always needed for diagnosis. Skin biopsy is a test in which doctors remove a thin skin sample and examine nerve fiber endings. This test offers some unique advantages over NCV tests and nerve biopsy. Unlike NCV, it can reveal damage present in smaller fibers; in contrast to conventional nerve biopsy, skin biopsy is less invasive, has fewer side effects, and is easier to perform. What treatments are available?No medical treatments now exist that can cure inherited peripheral neuropathy. However, there are therapies for many other forms. Any underlying condition is treated first, followed by symptomatic treatment. Peripheral nerves have the ability to regenerate, as long as the nerve cell itself has not been killed. Symptoms often can be controlled, and eliminating the causes of specific forms of neuropathy often can prevent new damage. In general, adopting healthy habits-such as maintaining optimal weight, avoiding exposure to toxins, following a physician-supervised exercise program, eating a balanced diet, correcting vitamin deficiencies, and limiting or avoiding alcohol consumption-can reduce the physical and emotional effects of peripheral neuropathy. Active and passive forms of exercise can reduce cramps, improve muscle strength, and prevent muscle wasting in paralyzed limbs. Various dietary strategies can improve gastrointestinal symptoms. Timely treatment of injury can help prevent permanent damage. Quitting smoking is particularly important because smoking constricts the blood vessels that supply nutrients to the peripheral nerves and can worsen neuropathic symptoms. Self-care skills such as meticulous foot care and careful wound treatment in people with diabetes and others who have an impaired ability to feel pain can alleviate symptoms and improve quality of life. Such changes often create conditions that encourage nerve regeneration. Systemic diseases frequently require more complex treatments. Strict control of blood glucose levels has been shown to reduce neuropathic symptoms and help people with diabetic neuropathy avoid further nerve damage. Inflammatory and autoimmune conditions leading to neuropathy can be controlled in several ways. Immunosuppressive drugs such as prednisone, cyclosporine, or azathioprine may be beneficial. Plasmapheresis-a procedure in which blood is removed, cleansed of immune system cells and antibodies, and then returned to the body-can limit inflammation or suppress immune system activity. High doses of immunoglobulins, proteins that function as antibodies, also can suppress abnormal immune system activity. Neuropathic pain is often difficult to control. Mild pain may sometimes be alleviated by analgesics sold over the counter. Several classes of drugs have recently proved helpful to many patients suffering from more severe forms of chronic neuropathic pain. These include mexiletine, a drug developed to correct irregular heart rhythms (sometimes associated with severe side effects); several antiepileptic drugs, including gabapentin, phenytoin, and carbamazepine; and some classes of antidepressants, including tricyclics such as amitriptyline. Injections of local anesthetics such as lidocaine or topical patches containing lidocaine may relieve more intractable pain. In the most severe cases, doctors can surgically destroy nerves; however, the results are often temporary and the procedure can lead to complications. Mechanical aids can help reduce pain and lessen the impact of physical disability. Hand or foot braces can compensate for muscle weakness or alleviate nerve compression. Orthopedic shoes can improve gait disturbances and help prevent foot injuries in people with a loss of pain sensation. If breathing becomes severely impaired, mechanical ventilation can provide essential life support. Surgical intervention often can provide immediate relief from mononeuropathies caused by compression or entrapment injuries. Repair of a slipped disk can reduce pressure on nerves where they emerge from the spinal cord; the removal of benign or malignant tumors can also alleviate damaging pressure on nerves. Nerve entrapment often can be corrected by the surgical release of ligaments or tendons. What research is being done?The National Institute of Neurological Disorders and Stroke (NINDS), a component of the Federal government's National Institutes of Health (NIH) within the U.S. Department of Health and Human Services, has primary responsibility for research on peripheral neuropathy. Current research projects funded by the NINDS involve investigations of genetic factors associated with hereditary neuropathies, studies of biological mechanisms involved in diabetes-associated neuropathies, efforts to gain greater understanding of how the immune system contributes to peripheral nerve damage, and efforts to develop new therapies for neuropathic symptoms. Because specific genetic defects have been identified for only a fraction of the known hereditary neuropathies, the Institute sponsors studies to identify other genetic defects that may cause these conditions. Presymptomatic diagnosis may lead to therapies for preventing nerve damage before it occurs, and gene replacement therapies could be developed to prevent or reduce cumulative nerve damage. Several NINDS-funded studies are investigating some of the possible biological mechanisms responsible for the many forms of neuropathy, including the autonomic neuropathies that affect people with diabetes. The Institute also is funding studies to measure the frequency and progression rates of diabetic neuropathies, examine the effects of these disorders on quality of life, and identify factors that may put certain individuals at greater risk for developing diabetes-associated neuropathies. Scientists have found that the destructive effects of abnormal immune system activity cause many neuropathies for which a cause could not previously be identified. However, the exact biological mechanisms that lead to this nerve damage are not yet well understood. Many NINDS-sponsored studies are studying inflammatory neuropathies, both in research animals and in humans, to clarify these mechanisms so that therapeutic interventions can be developed. Neuropathic pain is a primary target of NINDS-sponsored studies aimed at developing more effective therapies for symptoms of peripheral neuropathy. Some scientists hope to identify substances that will block the brain chemicals that generate pain signals, while others are investigating the pathways by which pain signals reach the brain. Studies of neurotrophic factors represent one of the most promising areas of research aimed at finding new, more effective treatments for peripheral neuropathies. These substances, produced naturally by the body, protect neurons from injury and encourage their survival. Neurotrophic factors also help maintain normal function in mature nerve cells, and some stimulate axon regeneration. Several NINDS-sponsored studies seek to learn more about the effects of these powerful chemicals on the peripheral nervous system and may eventually lead to treatments that can reverse nerve damage and cure peripheral nerve disorders. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 04-4853 |
| Parkinson's
Disease Introduction
What is Parkinson's Disease? What Causes the Disease? What Genes are Linked to Parkinson's Disease? Who Gets Parkinson's Disease? What are the Symptoms of the Disease? What Other Diseases Resemble Parkinson's? How is Parkinson's Disease Diagnosed? What is the Prognosis? How is the Disease Treated? Drug Treatments Surgery Complementary and Supportive Therapies How Can People Cope with Parkinson's Disease? Can Scientists Predict or Prevent Parkinson's Disease? What Research is Being Done? What is the Role of the NINDS? What Can I Do to Help? Where can I get more information? Glossary IntroductionParkinson's
disease (PD) is a degenerative disorder of the central nervous system. It
was first described in 1817 by James Parkinson, a British physician
who published a paper on what he called "the shaking palsy." In this
paper, he set forth the major symptoms of the disease that would
later bear his name.
Researchers believe that at least 500,000 people
in the
What is Parkinson's Disease?Parkinson's disease belongs to a group of conditions called movement disorders. The four main symptoms are tremor, or trembling in hands, arms, legs, jaw, or head; rigidity, or stiffness of the limbs and trunk; bradykinesia, or slowness of movement; and postural instability, or impaired balance. These symptoms usually begin gradually and worsen with time. As they become more pronounced, patients may have difficulty walking, talking, or completing other simple tasks. Not everyone with one or more of these symptoms has PD, as the symptoms sometimes appear in other diseases as well. PD is both chronic, meaning it persists over a long period of time, and progressive, meaning its symptoms grow worse over time. It is not contagious. Although some PD cases appear to be hereditary, and a few can be traced to specific genetic mutations, most cases are sporadic — that is, the disease does not seem to run in families. Many researchers now believe that PD results from a combination of genetic susceptibility and exposure to one or more environmental factors that trigger the disease. PD is the most common form of parkinsonism, the name for a group of disorders with similar features and symptoms. PD is also called primary parkinsonism or idiopathic PD. The term idiopathic means a disorder for which no cause has yet been found. While most forms of parkinsonism are idiopathic, there are some cases where the cause is known or suspected or where the symptoms result from another disorder. For example, parkinsonism may result from changes in the brain's blood vessels. What Causes the Disease?Parkinson's disease occurs when nerve cells, or neurons, in an area of the brain known as the substantia nigra die or become impaired. Normally, these neurons produce an important brain chemical known as dopamine. Dopamine is a chemical messenger responsible for transmitting signals between the substantia nigra and the next "relay station" of the brain, the corpus striatum, to produce smooth, purposeful movement. Loss of dopamine results in abnormal nerve firing patterns within the brain that cause impaired movement. Studies have shown that most Parkinson's patients have lost 60 to 80 percent or more of the dopamine-producing cells in the substantia nigra by the time symptoms appear. Recent studies have shown that people with PD also have loss of the nerve endings that produce the neurotransmitter norepinephrine. Norepinephrine, which is closely related to dopamine, is the main chemical messenger of the sympathetic nervous system, the part of the nervous system that controls many automatic functions of the body, such as pulse and blood pressure. The loss of norepinephrine might help explain several of the non-motor features seen in PD, including fatigue and abnormalities of blood pressure regulation. Many brain cells of people with PD contain Lewy bodies – unusual deposits or clumps of the protein alpha-synuclein, along with other proteins. Researchers do not yet know why Lewy bodies form or what role they play in development of the disease. The clumps may prevent the cell from functioning normally, or they may actually be helpful, perhaps by keeping harmful proteins "locked up" so that the cells can function. Scientists have identified several genetic mutations associated with PD, and many more genes have been tentatively linked to the disorder. Studying the genes responsible for inherited cases of PD can help researchers understand both inherited and sporadic cases. The same genes and proteins that are altered in inherited cases may also be altered in sporadic cases by environmental toxins or other factors. Researchers also hope that discovering genes will help identify new ways of treating PD. Although the importance of genetics in PD is increasingly recognized, most researchers believe environmental exposures increase a person's risk of developing the disease. Even in familial cases, exposure to toxins or other environmental factors may influence when symptoms of the disease appear or how the disease progresses. There are a number of toxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, or MPTP (found in some kinds of synthetic heroin), that can cause parkinsonian symptoms in humans. Other, still-unidentified environmental factors also may cause PD in genetically susceptible individuals. Viruses are another possible environmental trigger for PD. People who developed encephalopathy after a 1918 influenza epidemic were later stricken with severe, progressive Parkinson's-like symptoms. A group of Taiwanese women developed similar symptoms after contracting herpes virus infections. In these women, the symptoms, which later disappeared, were linked to a temporary inflammation of the substantia nigra. Several lines of research suggest that mitochondria may play a role in the development of PD. Mitochondria are the energy-producing components of the cell and are major sources of free radicals — molecules that damage membranes, proteins, DNA, and other parts of the cell. This damage is often referred to as oxidative stress. Oxidative stress-related changes, including free radical damage to DNA, proteins, and fats, have been detected in brains of PD patients. Other research suggests that the cell's protein disposal system may fail in people with PD, causing proteins to build up to harmful levels and trigger cell death. Additional studies have found evidence that clumps of protein that develop inside brain cells of people with PD may contribute to the death of neurons, and that inflammation or overstimulation of cells (because of toxins or other factors) may play a role in the disease. However, the precise role of the protein deposits remains unknown. Some researchers even speculate that the protein buildup is part of an unsuccessful attempt to protect the cell. While mitochondrial dysfunction, oxidative stress, inflammation, and many other cellular processes may contribute to PD, the actual cause of the dopamine cell death is still undetermined. What Genes are Linked to Parkinson's Disease?Several
genes have now been definitively linked to PD. The
first to be identified was alpha-synuclein. In the
1990s, researchers at NIH and other institutions studied
the genetic profiles of a large Italian family and
three Greek families with familial PD and found that
their disease was related to a mutation in this gene.
They found a second alpha-synuclein mutation in a German
family with PD. These findings prompted studies of
the role of alpha-synuclein in PD, which led to the
discovery that Lewy bodies from people with the sporadic
form of PD contained clumps of alpha-synuclein protein.
This discovery revealed a potential link between hereditary
and sporadic forms of the disease.
In 2003, researchers studying inherited PD discovered that the disease in one large family was caused by a triplication of the normal alpha-synuclein gene on one copy of chromosome 4. This triplication caused people in the affected family to produce too much of the normal alpha-synuclein. This study showed that an excess of the normal form of the protein could result in PD, just as the abnormal form does. Other genes linked to PD include parkin, DJ-1, PINK1, and LRRK2. Parkin, DJ-1, and PINK-1 cause rare, early-onset forms of PD. The parkin gene is translated into a protein that normally helps cells break down and recycle proteins. DJ-1 normally helps regulate gene activity and protect cells from oxidative stress. PINK1 codes for a protein active in mitochondria. Mutations in this gene appear to increase susceptibility to cellular stress. LRRK2, which is translated into a protein called dardarin, was originally identified in several English and Basque families and causes a late-onset form of PD. Subsequent studies have identified this gene in other families with PD as well as in a small percentage of people with apparently sporadic PD. Researchers are continuing to investigate the normal functions and interactions of these genes in order to find clues about how PD develops. They also have identified a number of other genes and chromosome regions that may play a role in PD, but the nature of these links is not yet clear. Who Gets Parkinson's Disease?About 50,000 Americans are diagnosed with PD each year, but getting an accurate count of the number of cases may be impossible because many people in the early stages of the disease assume their symptoms are the result of normal aging and do not seek help from a physician. Also, diagnosis is sometimes difficult and uncertain because other conditions may produce symptoms of PD and there is no definitive test for the disease. People with PD may sometimes be told by their doctors that they have other disorders, and people with PD-like diseases may be incorrectly diagnosed as having PD. PD strikes about 50 percent more men than women, but the reasons for this discrepancy are unclear. While it occurs in people throughout the world, a number of studies have found a higher incidence in developed countries, possibly because of increased exposure to pesticides or other toxins in those countries. Other studies have found an increased risk in people who live in rural areas and in those who work in certain professions, although the studies to date are not conclusive and the reasons for the apparent risks are not clear. One clear risk factor for PD is age. The average age of onset is 60 years, and the incidence rises significantly with increasing age. However, about 5 to 10 percent of people with PD have "early-onset" disease that begins before the age of 50. Early-onset forms of the disease are often inherited, though not always, and some have been linked to specific gene mutations. People with one or more close relatives who have PD have an increased risk of developing the disease themselves, but the total risk is still just 2 to 5 percent unless the family has a known gene mutation for the disease. An estimated 15 to 25 percent of people with PD have a known relative with the disease. In
very rare cases, parkinsonian symptoms may appear
in people before the age of 20. This condition
is called juvenile parkinsonism.
It is most commonly seen in
What are the Symptoms of the Disease?Early symptoms of PD are subtle and occur gradually. Affected people may feel mild tremors or have difficulty getting out of a chair. They may notice that they speak too softly or that their handwriting is slow and looks cramped or small. They may lose track of a word or thought, or they may feel tired, irritable, or depressed for no apparent reason. This very early period may last a long time before the more classic and obvious symptoms appear. Friends or family members may be the first to notice changes in someone with early PD. They may see that the person's face lacks expression and animation (known as "masked face") or that the person does not move an arm or leg normally. They also may notice that the person seems stiff, unsteady, or unusually slow. As the disease progresses, the shaking or tremor that affects the majority of Parkinson's patients may begin to interfere with daily activities. Patients may not be able to hold utensils steady or they may find that the shaking makes reading a newspaper difficult. Tremor is usually the symptom that causes people to seek medical help. People with PD often develop a so-called parkinsonian gait that includes a tendency to lean forward, small quick steps as if hurrying forward (called festination), and reduced swinging of the arms. They also may have trouble initiating movement (start hesitation), and they may stop suddenly as they walk (freezing). PD does not affect everyone the same way, and the rate of progression differs among patients. Tremor is the major symptom for some patients, while for others, tremor is nonexistent or very minor. PD symptoms often begin on one side of the body. However, as it progresses, the disease eventually affects both sides. Even after the disease involves both sides of the body, the symptoms are often less severe on one side than on the other. The four primary symptoms of PD are:
A number of other symptoms may accompany PD. Some are minor; others are not. Many can be treated with medication or physical therapy. No one can predict which symptoms will affect an individual patient, and the intensity of the symptoms varies from person to person.
What Other Diseases Resemble Parkinson's?A number of disorders can cause symptoms similar to those of PD. People with symptoms that resemble PD but that result from other causes are sometimes said to have parkinsonism. Some of these disorders are listed below.
MSA, corticobasal degeneration, and progressive supranuclear palsy are sometimes referred to as "Parkinson's-plus" diseases because they have the symptoms of PD plus additional features. How is Parkinson's Disease Diagnosed?There are currently no blood or laboratory tests that have been proven to help in diagnosing sporadic PD. Therefore the diagnosis is based on medical history and a neurological examination. The disease can be difficult to diagnose accurately. Early signs and symptoms of PD may sometimes be dismissed as the effects of normal aging. The physician may need to observe the person for some time until it is apparent that the symptoms are consistently present. Doctors may sometimes request brain scans or laboratory tests in order to rule out other diseases. However, CT and MRI brain scans of people with PD usually appear normal. Since many other diseases have similar features but require different treatments, making a precise diagnosis as soon as possible is essential so that patients can receive the proper treatment. What is the Prognosis?PD is not by itself a fatal disease, but it does get worse with time. The average life expectancy of a PD patient is generally the same as for people who do not have the disease. However, in the late stages of the disease, PD may cause complications such as choking, pneumonia, and falls that can lead to death. Fortunately, there are many treatment options available for people with PD. The progression of symptoms in PD may take 20 years or more. In some people, however, the disease progresses more quickly. There is no way to predict what course the disease will take for an individual person. One commonly used system for describing how the symptoms of PD progress is called the Hoehn and Yahr scale. Hoehn and Yahr Staging of Parkinson's Disease
Another commonly used scale is the Unified Parkinson's Disease Rating Scale (UPDRS). This much more complicated scale has multiple ratings that measure mental functioning, behavior, and mood; activities of daily living; and motor function. Both the Hoehn and Yahr scale and the UPDRS are used to measure how individuals are faring and how much treatments are helping them. With appropriate treatment, most people with PD can live productive lives for many years after diagnosis. How is the Disease Treated?At present, there is no cure for PD. But medications or surgery can sometimes provide dramatic relief from the symptoms. Drug TreatmentsMedications for PD fall into three categories. The first category includes drugs that work directly or indirectly to increase the level of dopamine in the brain. The most common drugs for PD are dopamine precursors – substances such as levodopa that cross the blood-brain barrier and are then changed into dopamine. Other drugs mimic dopamine or prevent or slow its breakdown. The second category of PD drugs affects other neurotransmitters in the body in order to ease some of the symptoms of the disease. For example, anticholinergic drugs interfere with production or uptake of the neurotransmitter acetylcholine. These drugs help to reduce tremors and muscle stiffness, which can result from having more acetylcholine than dopamine. The third category of drugs prescribed for PD includes medications that help control the non-motor symptoms of the disease, that is, the symptoms that don't affect movement. For example, people with PD-related depression may be prescribed antidepressants.
Fortunately, physicians have other treatment choices for some symptoms and stages of PD. These therapies include the following:
When recommending a course of treatment, a doctor will assess how much the symptoms disrupt the patient's life and then tailor therapy to the person's particular condition. Since no two patients will react the same way to a given drug, it may take time and patience to get the dose just right. Even then, symptoms may not be completely alleviated. Medications to Treat the Motor Symptoms of Parkinson's Disease
Medications for Non-Motor Symptoms. Doctors may prescribe a variety of medications to treat the non-motor symptoms of PD, such as depression and anxiety. For example, depression can be treated with standard anti-depressant drugs such as amitriptyline or fluoxetine (however, as stated earlier, fluoxetine should not be combined with MAO-B inhibitors). Anxiety can sometimes be treated with drugs called benzodiazepines. Orthostatic hypotension may be helped by increasing salt intake, reducing antihypertension drugs, or prescribing medications such as fludrocortisone. Hallucinations, delusions, and other psychotic symptoms are often caused by the drugs prescribed for PD. Therefore reducing or stopping PD medications may alleviate psychosis. If such measures are not effective, doctors sometimes prescribe drugs called atypical antipsychotics, which include clozapine and quetiapine. Clozapine also may help to control dyskinesias. However, clozapine also can cause a serious blood disorder called agranulocytosis, so people who take it must have their blood monitored frequently. SurgeryTreating PD with surgery was once a common practice. But after the discovery of levodopa, surgery was restricted to only a few cases. Studies in the past few decades have led to great improvements in surgical techniques, and surgery is again being used in people with advanced PD for whom drug therapy is no longer sufficient. Pallidotomy and Thalamotomy. The earliest types of surgery for PD involved selectively destroying specific parts of the brain that contribute to the symptoms of the disease. Investigators have now greatly refined the use of these procedures. The most common of these procedures is called pallidotomy. In this procedure, a surgeon selectively destroys a portion of the brain called the globus pallidus. Pallidotomy can improve symptoms of tremor, rigidity, and bradykinesia, possibly by interrupting the connections between the globus pallidus and the striatum or thalamus. Some studies have also found that pallidotomy can improve gait and balance and reduce the amount of levodopa patients require, thus reducing drug-induced dyskinesias and dystonia. A related procedure, called thalamotomy, involves surgically destroying part of the brain's thalamus. Thalamotomy is useful primarily to reduce tremor. Because these procedures cause permanent destruction of brain tissue, they have largely been replaced by deep brain stimulation for treatment of PD. Deep Brain Stimulation. Deep brain stimulation, or DBS, uses an electrode surgically implanted into part of the brain. The electrodes are connected by a wire under the skin to a small electrical device called a pulse generator that is implanted in the chest beneath the collarbone. The pulse generator and electrodes painlessly stimulate the brain in a way that helps to stop many of the symptoms of PD. DBS has now been approved by the U.S. Food and Drug Administration, and it is widely used as a treatment for PD. DBS can be used on one or both sides of the brain. If it is used on just one side, it will affect symptoms on the opposite side of the body. DBS is primarily used to stimulate one of three brain regions: the subthalamic nucleus, the globus pallidus, or the thalamus. However, the subthalamic nucleus, a tiny area located beneath the thalamus, is the most common target. Stimulation of either the globus pallidus or the subthalamic nucleus can reduce tremor, bradykinesia, and rigidity. Stimulation of the thalamus is useful primarily for reducing tremor. DBS usually reduces the need for levodopa and related drugs, which in turn decreases dyskinesias. It also helps to relieve on-off fluctuation of symptoms. People who initially responded well to treatment with levodopa tend to respond well to DBS. While the benefits of DBS can be substantial, it usually does not help with speech problems, "freezing," posture, balance, anxiety, depression, or dementia. One advantage of DBS compared to pallidotomy and thalamotomy is that the electrical current can be turned off using a handheld device. The pulse generator also can be externally programmed. Patients must return to the medical center frequently for several months after DBS surgery in order to have the stimulation adjusted by trained doctors or other medical professionals. The pulse generator must be programmed very carefully to give the best results. Doctors also must supervise reductions in patients' medications. After a few months, the number of medical visits usually decreases significantly, though patients may occasionally need to return to the center to have their stimulator checked. Also, the battery for the pulse generator must be surgically replaced every three to five years, though externally rechargeable batteries may eventually become available. Long-term results of DBS are still being determined. DBS does not stop PD from progressing, and some problems may gradually return. However, studies up to several years after surgery have shown that many people's symptoms remain significantly better than they were before DBS. DBS is not a good solution for everyone. It is generally used only in people with advanced, levodopa-responsive PD who have developed dyskinesias or other disabling "off" symptoms despite drug therapy. It is not normally used in people with memory problems, hallucinations, a poor response to levodopa, severe depression, or poor health. DBS generally does not help people with "atypical" parkinsonian syndromes such as multiple system atrophy, progressive supranuclear palsy, or post-traumatic parkinsonism. Younger people generally do better than older people after DBS, but healthy older people can undergo DBS and they may benefit a great deal. As with any brain surgery, DBS has potential complications, including stroke or brain hemorrhage. These complications are rare, however. There is also a risk of infection, which may require antibiotics or even replacement of parts of the DBS system. The stimulator may sometimes cause speech problems, balance problems, or even dyskinesias. However, those problems are often reversible if the stimulation is modified. Researchers are continuing to study DBS and to develop ways of improving it. They are conducting clinical studies to determine the best part of the brain to receive stimulation and to determine the long-term effects of this therapy. They also are working to improve the technology used in DBS. Complementary and Supportive TherapiesA wide variety of complementary and supportive therapies may be used for PD. Among these therapies are standard physical, occupational, and speech therapy techniques, which can help with such problems as gait and voice disorders, tremors and rigidity, and cognitive decline. Other types of supportive therapies include the following: Diet. At this time there are no specific vitamins, minerals, or other nutrients that have any proven therapeutic value in PD. Some early reports have suggested that dietary supplements might be protective in PD. In addition, a phase II clinical trial of a supplement called coenzyme Q10 suggested that large doses of this substance might slow disease progression in patients with early-stage PD. The NINDS and other components of the National Institutes of Health are funding research to determine if caffeine, antioxidants, and other dietary factors may be beneficial for preventing or treating PD. While there is currently no proof that any specific dietary factor is beneficial, a normal, healthy diet can promote overall well-being for PD patients just as it would for anyone else. Eating a fiber-rich diet and drinking plenty of fluids also can help alleviate constipation. A high protein diet, however, may limit levodopa's effectiveness. Exercise. Exercise can help people with PD improve their mobility and flexibility. Some doctors prescribe physical therapy or muscle-strengthening exercises to tone muscles and to put underused and rigid muscles through a full range of motion. Exercises will not stop disease progression, but they may improve body strength so that the person is less disabled. Exercises also improve balance, helping people minimize gait problems, and can strengthen certain muscles so that people can speak and swallow better. Exercise can also improve the emotional well-being of people with PD, and it may improve the brain's dopamine synthesis or increase levels of beneficial compounds called neurotrophic factors in the brain. Although structured exercise programs help many patients, more general physical activity, such as walking, gardening, swimming, calisthenics, and using exercise machines, also is beneficial. People with PD should always check with their doctors before beginning a new exercise program. Other complementary therapies that are used by some individuals with PD include massage therapy, yoga, tai chi, hypnosis, acupuncture, and the Alexander technique, which optimizes posture and muscle activity. There have been limited studies suggesting mild benefits with some of these therapies, but they do not slow PD and there is no convincing evidence that they are beneficial. How Can People Cope with Parkinson's Disease?While PD usually progresses slowly, eventually the most basic daily routines may be affected — from socializing with friends and enjoying normal relationships with family members to earning a living and taking care of a home. These changes can be difficult to accept. Support groups can help people cope with the disease emotionally. These groups can also provide valuable information, advice, and experience to help people with PD, their families, and their caregivers deal with a wide range of issues, including locating doctors familiar with the disease and coping with physical limitations. A list of national organizations that can help patients locate support groups in their communities appears at the end of this brochure. Individual or family counseling also may help people find ways to cope with PD. People with PD also can benefit from being proactive and finding out as much as possible about the disease in order to alleviate fear of the unknown and to take a positive role in maintaining their health. Many people with PD continue to work either full- or part-time, although eventually they may need to adjust their schedule and working environment to cope with the disease. Can Scientists Predict or Prevent Parkinson's Disease?In most cases, there is no way to predict or prevent sporadic PD. However, researchers are looking for a biomarker — a biochemical abnormality that all patients with PD might share — that could be picked up by screening techniques or by a simple chemical test given to people who do not have any parkinsonian symptoms. This could help doctors identify people at risk of the disease. It also might allow them to find treatments that will stop the disease process in the early stages. Positron emission tomography (PET) scanning may lead to important advances in our knowledge about PD. PET scans of the brain produce pictures of chemical changes as they occur. Using PET, research scientists can study the brain's dopamine receptors (the sites on nerve cells that bind with dopamine) to determine if the loss of dopamine activity follows or precedes degeneration of the neurons that make this chemical. This information could help scientists better understand the disease process and may potentially lead to improved treatments. In rare cases, where people have a clearly inherited form of PD, researchers can test for known gene mutations as a way of determining an individual's risk of the disease. However, this genetic testing can have far-reaching implications and people should carefully consider whether they want to know the results of such tests. Genetic testing is currently available only as a part of research studies. What Research is Being Done?In recent years, Parkinson's research has advanced to the point that halting the progression of PD, restoring lost function, and even preventing the disease are all considered realistic goals. While the ultimate goal of preventing PD may take years to achieve, researchers are making great progress in understanding and treating PD. One of the most exciting areas of PD research is genetics. Studying the genes responsible for inherited cases can help researchers understand both inherited and sporadic cases of the disease. Identifying gene defects can also help researchers understand how PD occurs, develop animal models that accurately mimic the neuronal death in human PD, identify new drug targets, and improve diagnosis. As discussed in the “What Genes are Linked to Parkinson's Disease?" section, several genes have been definitively linked to PD in some people. Researchers also have identified a number of other genes that may play a role and are working to confirm these findings. In addition, several chromosomal regions have been linked to PD in some families. Researchers hope to identify the genes located in these chromosomal regions and to determine which of them may play roles in PD. Researchers funded by NINDS are gathering information and DNA samples from hundreds of families with PD and are conducting large-scale gene expression studies to identify genes that are abnormally active or inactive in PD. They also are comparing gene activity in PD with gene activity in similar diseases such as progressive supranuclear palsy. Some scientists have found evidence that specific variations in the DNA of mitochondria – structures in cells that provide the energy for cellular activity — can increase the risk of getting PD, while other variations are associated with a lowered risk of the disorder. They also have found that PD patients have more mitochondrial DNA (mtDNA) variations than patients with other movement disorders or Alzheimer's disease. Researchers are working to define how these mtDNA variations may lead to PD. In addition to identifying new genes for PD, researchers are trying to learn how known PD genes function and how the gene mutations cause disease. For example, a 2005 study found that the normal alpha-synuclein protein may help other proteins that are important for nerve transmission to fold correctly. Other studies have suggested that the normal parkin protein protects neurons from a variety of threats, including alpha-synuclein toxicity and excitotoxicity. Scientists continue to study environmental toxins such as pesticides and herbicides that can cause PD symptoms in animals. They have found that exposing rodents to the pesticide rotenone and several other agricultural chemicals can cause cellular and behavioral changes that mimic those seen in PD. Other studies have suggested that prenatal exposure to certain toxins can increase susceptibility to PD in adulthood. An NIH-sponsored program called the Collaborative Centers for Parkinson's Disease Environmental Research (CCPDER) focuses on how occupational exposure to toxins and use of caffeine and other substances may affect the risk of PD. Another major area of PD research involves the cell's protein disposal system, called the ubiquitin-proteasome system. If this disposal system fails to work correctly, toxins and other substances may build up to harmful levels, leading to cell death. The ubiquitin-proteasome system requires interactions between several proteins, including parkin and UCH-L1. Therefore, disruption of the ubiquitin-proteasome system may partially explain how mutations in these genes cause PD. Other studies focus on how Lewy bodies form and what role they play in PD. Some studies suggest that Lewy bodies are a byproduct of degenerative processes within neurons, while others indicate that Lewy bodies are a protective mechanism by which neurons lock away abnormal molecules that might otherwise be harmful. Additional studies have found that alpha-synuclein clumps alter gene expression and bind to vesicles within the cell in ways that could be harmful. Another common topic of PD research is excitotoxicity – overstimulation of nerve cells that leads to cell damage or death. In excitotoxicity, the brain becomes oversensitized to the neurotransmitter glutamate, which increases activity in the brain. The dopamine deficiency in PD causes overactivity of neurons in the subthalamic nucleus, which may lead to excitotoxic damage there and in other parts of the brain. Researchers also have found that dysfunction of the cells' mitochondria can make dopamine-producing neurons vulnerable to glutamate. Other researchers are focusing on how inflammation may affect PD. Inflammation is common to a variety of neurodegenerative diseases, including PD, Alzheimer's disease, HIV-1-associated dementia, and amyotrophic lateral sclerosis. Several studies have shown that inflammation-promoting molecules increase cell death after treatment with the toxin MPTP. Inhibiting the inflammation with drugs or by genetic engineering prevented some of the neuronal degeneration in these studies. Other research has shown that dopamine neurons in brains from patients with PD have higher levels of an inflammatory enzyme called COX-2 than those of people without PD. Inhibiting COX-2 doubled the number of neurons that survived in a mouse model for PD. Since the discovery that MPTP causes parkinsonian symptoms in humans, scientists have found that by injecting MPTP and certain other toxins into laboratory animals, they can reproduce the brain lesions that cause these symptoms. This allows them to study the mechanisms of the disease and helps in the development of new treatments. They also have developed animal models with alterations of the alpha-synuclein and parkin genes. Other researchers have used genetic engineering to develop mice with disrupted mitochondrial function in dopamine neurons. These animals have many of the characteristics associated with PD. Biomarkers for PD – measurable characteristics that can reveal whether the disease is developing or progressing – are another focus of research. Such biomarkers could help doctors detect the disease before symptoms appear and improve diagnosis of the disease. They also would show if medications and other types of therapy have a positive or negative effect on the course of the disease. Some of the most promising biomarkers for PD are brain imaging techniques. For example, some researchers are using positron emission tomography (PET) brain scans to try to identify metabolic changes in the brains of people with PD and to determine how these changes relate to disease symptoms. Other potential biomarkers for PD include alterations in gene expression. Researchers also are conducting many studies of new or improved therapies for PD. While deep brain stimulation (DBS) is now FDA-approved and has been used in thousands of people with PD, researchers continue to try to improve the technology and surgical techniques in this therapy. For example, some studies are comparing DBS to the best medical therapy and trying to determine which part of the brain is the best location for stimulation. Another clinical trial is studying how DBS affects depression and quality of life. Other clinical studies are testing whether transcranial electrical polarization (TEP) or transcranial magnetic stimulation (TMS) can reduce the symptoms of PD. In TEP, electrodes placed on the scalp are used to generate an electrical current that modifies signals in the brain's cortex. In TMS, an insulated coil of wire on the scalp is used to generate a brief electrical current. One of the enduring questions in PD research has been how treatment with levodopa and other dopaminergic drugs affects progression of the disease. Researchers are continuing to try to clarify these effects. One study has suggested that PD patients with a low-activity variant of the gene for COMT (which breaks down dopamine) perform worse than others on tests of cognition, and that dopaminergic drugs may worsen cognition in these people, perhaps because the reduced COMT activity causes dopamine to build up to harmful levels in some parts of the brain. In the future, it may become possible to test for such individual gene differences in order to improve treatment of PD. A variety of new drug treatments are in clinical trials for PD. These include a drug called GM1 ganglioside that increases dopamine levels in the brain. Researchers are testing whether this drug can reduce symptoms, delay disease progression, or partially restore damaged brain cells in PD patients. Other studies are testing whether a drug called istradefylline can improve motor function in PD, and whether a drug called ACP-103 that blocks receptors for the neurotransmitter serotonin will lessen the severity of parkinsonian symptoms and levodopa-associated complications in PD patients. Other topics of research include controlled-release formulas of PD drugs and implantable pumps that give a continuous supply of levodopa. Some researchers are testing potential neuroprotective drugs to see if they can slow the progression of PD. One study, called NET-PD (Neuroexploratory Trials in Parkinson's Disease), is evaluating minocycline, creatine, coenzyme Q10, and GPI-1485 to determine if any of these agents should be considered for further testing. The NET-PD study may evaluate other possible neuroprotective agents in the future. Drugs found to be successful in the pilot phases may move to large phase III trials involving hundreds of patients. A separate group of researchers is investigating the effects of either 1200 or 2400 milligrams of coenzyme Q10 in 600 patients. Several MAO-B inhibitors, including selegiline, lazabemide, and rasagiline, also are in clinical trials to determine if they have neuroprotective effects in people with PD. Nerve growth factors, or neurotrophic factors, which support survival, growth, and development of brain cells, are another type of potential therapy for PD. One such drug, glial cell line-derived neurotrophic factor (GDNF), has been shown to protect dopamine neurons and to promote their survival in animal models of PD. This drug has been tested in several clinical trials for people with PD, and the drug appeared to cause regrowth of dopamine nerve fibers in one person who received the drug. However, a phase II clinical study of GDNF was halted in 2004 because the treatment did not show any clinical benefit after 6 months, and some data suggested that it might even be harmful. Other neurotrophins that may be useful for treating PD include neurotrophin-4 (NT-4), brain-derived neurotrophic factor (BDNF), and fibroblast growth factor 2 (FGF-2). While there is currently no proof that any dietary supplements can slow PD, several clinical studies are testing whether supplementation with vitamin B12 and other substances may be helpful. A 2005 study found that dietary restriction — reducing the number of calories normally consumed – helped to increase abnormally low levels of the neurotransmitter glutamate in a mouse model for early PD. The study also suggested that dietary restriction affected dopamine activity in the brain. Another study showed that dietary restriction before the onset of PD in a mouse model helped to protect dopamine-producing neurons. Other studies are looking at treatments that might improve some of the secondary symptoms of PD, such as depression and swallowing disorders. One clinical trial is investigating whether a drug called quetiapine can reduce psychosis or agitation in PD patients with dementia and in dementia patients with parkinsonian symptoms. Some studies also are examining whether transcranial magnetic stimulation or a food supplement called s-adenosyl-methionine (SAM-e) can alleviate depression in people with PD, and whether levetiracetam, a drug approved to treat epilepsy, can reduce dyskinesias in Parkinson's patients without interfering with other PD drugs. Another approach to treating PD is to implant cells to replace those lost in the disease. Researchers are conducting clinical trials of a cell therapy in which human retinal epithelial cells attached to microscopic gelatin beads are implanted into the brains of people with advanced PD. The retinal epithelial cells produce levodopa. The investigators hope that this therapy will enhance brain levels of dopamine. Starting in the 1990s, researchers conducted a controlled clinical trial of fetal tissue implants in people with PD. They attempted to replace lost dopamine-producing neurons with healthy ones from fetal tissue in order to improve movement and the response to medications. While many of the implanted cells survived in the brain and produced dopamine, this therapy was associated with only modest functional improvements, mostly in patients under the age of 60. Unfortunately, some of the people who received the transplants developed disabling dyskinesias that could not be relieved by reducing antiparkinsonian medications. Another type of cell therapy involves stem cells. Stem cells derived from embryos can develop into any kind of cell in the body, while others, called progenitor cells, are more restricted. One study transplanted neural progenitor cells derived from human embryonic stem cells into a rat model of PD. The cells appeared to trigger improvement on several behavioral tests, although relatively few of the transplanted cells became dopamine-producing neurons. Other researchers are developing methods to improve the number of dopamine-producing cells that can be grown from embryonic stem cells in culture. Researchers also are exploring whether stem cells from adult brains might be useful in treating PD. They have shown that the brain's white matter contains multipotent progenitor cells that can multiply and form all the major cell types of the brain, including neurons. Gene therapy is yet another approach to treating PD. A study of gene therapy in non-human primate models of PD is testing different genes and gene-delivery techniques in an effort to refine this kind of treatment. An early-phase clinical study is also testing whether using the adeno-associated virus type 2 (AAV2) to deliver the gene for a nerve growth factor called neurturin is safe for use in people with PD. Another study is testing the safety of gene therapy using AAV to deliver a gene for human aromatic L-amino acid decarboxylase, an enzyme that helps convert levodopa to dopamine in the brain. Other investigators are testing whether gene therapy to increase the amount of glutamic acid decarboxylase, which helps produce an inhibitory neurotransmitter called GABA, might reduce the overactivity of neurons in the brain that results from lack of dopamine. Another potential approach to treating PD is to use a vaccine to modify the immune system in a way that can protect dopamine-producing neurons. One vaccine study in mice used a drug called copolymer-1 that increases the number of immune T cells that secrete anti-inflammatory cytokines and growth factors. The researchers injected copolymer-1-treated immune cells into a mouse model for PD. The vaccine modified the behavior of supporting (glial) cells in the brain so that their responses were beneficial rather than harmful. It also reduced the amount of neurodegeneration in the mice, reduced inflammation, and increased production of nerve growth factors. Another study delivered a vaccine containing alpha-synuclein in a mouse model of PD and showed that the mice developed antibodies that reduced the accumulation of abnormal alpha-synuclein. While these studies are preliminary, investigators hope that similar approaches might one day be tested in humans. What is the Role of the NINDS?As
a world leader in research on neurological disorders,
including PD, the NINDS supports a wide range of
basic laboratory studies and clinical trials at its
The NINDS also supports 11 Morris K. Udall Parkinson’s Disease Research Centers of Excellence throughout the country. The Centers’ multidisciplinary research environment allows scientists to take advantage of new discoveries in the basic and technological sciences that could lead to clinical advances. Most of the Centers also provide state-of-the-art training for young scientists preparing for research careers investigating PD and related neurological disorders. Among other topics, the Centers carry out studies of genes, of proteins involved in cell death and degeneration, and of the brain chemicals involved in PD. They also study the brain using PET brain scans and test potential PD treatments in animals. The NINDS hopes that research at these Centers of Excellence will lead to clinical trials of new therapies in humans with PD. What Can I Do to Help?The NINDS and the National Institute of Mental Health jointly support two national brain specimen banks. These banks supply research scientists around the world with nervous system tissue from patients with neurological and psychiatric disorders. They need tissue from patients with PD so that scientists can study and understand the disorder. Those who may be interested in donating should contact: Rashed
M. Nagra, Ph.D., Director Francine
M. Benes, M.D., Ph.D., Director Two other organizations also provide research scientists with nervous system tissue from patients with neurological disorders. Interested donors should write or call: National
Disease Research Interchange UM/NPF
Brain Endowment Bank The Mohammed Ali Parkinson Center at the Barrow Neurological Institute in Phoenix, Arizona, has developed a national registry of people with PD in order to help in the development of new therapies and to allow researchers to quickly identify and notify people about research studies for which they are eligible. Anyone diagnosed with PD is eligible to take part in this registry. For more information, contact: Parkinson's
Disease Registry Some states, including California and Nebraska, also have registries of people with PD. People with PD who wish to help with research on this disorder may be able to do so by participating in clinical studies designed to learn more about the disease or to test potential new therapies. Information about many such studies is available free of charge from the Federal government's database of clinical trials, clinicaltrials.gov. A good source for finding clinical trials specifically on PD is the www.PDtrials.org web site, which lists studies sponsored by the National Institutes of Health and other federal agencies, as well as private industry and institutions at locations across the United States. This resource is sponsored by the Parkinson’s Disease Foundation in collaboration with the American Parkinson Disease Association, the Parkinson’s Action Network, the Parkinson Alliance, the Michael J. Fox Foundation for Parkinson’s Research, the National Parkinson Foundation, WE MOVE, and the NINDS. For clinical trials taking place at the National Institutes of Health, additional information is available from the following office: Patient
Recruitment and Public Liaison Office Where can I get more information? For more information on neurological disorders or research
programs funded by the National Institute of Neurological
Disorders and Stroke, contact the Institute's Brain Resources
and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 06-139 |
| Restless
Legs Syndrome What is restless
legs?
What are common signs and symptoms of restless legs? What causes restless legs syndrome? How is restless legs syndrome diagnosed? How is restless legs syndrome treated? What is the prognosis of people with restless legs? What research is being done? Where can I get more information? What is restless legs?Restless legs syndrome (RLS) is a neurological disorder characterized by unpleasant sensations in the legs and an uncontrollable urge to move when at rest in an effort to relieve these feelings. RLS sensations are often described by people as burning, creeping, tugging, or like insects crawling inside the legs. Often called paresthesias (abnormal sensations) or dysesthesias (unpleasant abnormal sensations), the sensations range in severity from uncomfortable to irritating to painful. The most distinctive or unusual aspect of the condition is that lying down and trying to relax activates the symptoms. As a result, most people with RLS have difficulty falling asleep and staying asleep. Left untreated, the condition causes exhaustion and daytime fatigue. Many people with RLS report that their job, personal relations, and activities of daily living are strongly affected as a result of their exhaustion. They are often unable to concentrate, have impaired memory, or fail to accomplish daily tasks. Some researchers estimate that RLS affects as many as 12 million Americans. However, others estimate a much higher occurrence because RLS is thought to be underdiagnosed and, in some cases, misdiagnosed. Some people with RLS will not seek medical attention, believing that they will not be taken seriously, that their symptoms are too mild, or that their condition is not treatable. Some physicians wrongly attribute the symptoms to nervousness, insomnia, stress, arthritis, muscle cramps, or aging. RLS occurs in both genders, although the incidence may be slightly higher in women. Although the syndrome may begin at any age, even as early as infancy, most patients who are severely affected are middle-aged or older. In addition, the severity of the disorder appears to increase with age. Older patients experience symptoms more frequently and for longer periods of time. More than 80 percent of people with RLS also experience a more common condition known as periodic limb movement disorder (PLMD). PLMD is characterized by involuntary leg twitching or jerking movements during sleep that typically occur every 10 to 60 seconds, sometimes throughout the night. The symptoms cause repeated awakening and severely disrupted sleep. Unlike RLS, the movements caused by PLMD are involuntary-people have no control over them. Although many patients with RLS also develop PLMD, most people with PLMD do not experience RLS. Like RLS, the cause of PLMD is unknown. What are common signs and symptoms of restless legs?As described above, people with RLS feel uncomfortable sensations in their legs, especially when sitting or lying down, accompanied by an irresistible urge to move about. These sensations usually occur deep inside the leg, between the knee and ankle; more rarely, they occur in the feet, thighs, arms, and hands. Although the sensations can occur on just one side of the body, they most often affect both sides. Because moving the legs (or other affected parts of the body) relieves the discomfort, people with RLS often keep their legs in motion to minimize or prevent the sensations. They may pace the floor, constantly move their legs while sitting, and toss and turn in bed. Most people find the symptoms to be less noticeable during the day and more pronounced in the evening or at night, especially during the onset of sleep. For many people, the symptoms disappear by early morning, allowing for more refreshing sleep at that time. Other triggering situations are periods of inactivity such as long car trips, sitting in a movie theater, long-distance flights, immobilization in a cast, or relaxation exercises. The symptoms of RLS vary in severity and duration from person to person. Mild RLS occurs episodically, with only mild disruption of sleep onset, and causes little distress. In moderately severe cases, symptoms occur only once or twice a week but result in significant delay of sleep onset, with some disruption of daytime function. In severe cases of RLS, the symptoms occur more than twice a week and result in burdensome interruption of sleep and impairment of daytime function. Symptoms may begin at any stage of life, although the disorder is more common with increasing age. Sometimes people will experience spontaneous improvement over a period of weeks or months. Although rare, spontaneous improvement over a period of years also can occur. If these improvements occur, it is usually during the early stages of the disorder. In general, however, symptoms become more severe over time. People who have both RLS and an associated condition tend to develop more severe symptoms rapidly. In contrast, those whose RLS is not related to any other medical condition and whose onset is at an early age show a very slow progression of the disorder and many years may pass before symptoms occur regularly. What causes restless legs syndrome?In most cases, the cause of RLS is unknown (referred to as idiopathic). A family history of the condition is seen in approximately 50 percent of such cases, suggesting a genetic form of the disorder. People with familial RLS tend to be younger when symptoms start and have a slower progression of the condition. In other cases, RLS appears to be related to the following factors or conditions, although researchers do not yet know if these factors actually cause RLS.
Researchers also have found that caffeine, alcohol, and tobacco may aggravate or trigger symptoms in patients who are predisposed to develop RLS. Some studies have shown that a reduction or complete elimination of such substances may relieve symptoms, although it remains unclear whether elimination of such substances can prevent RLS symptoms from occurring at all. How is restless legs syndrome diagnosed?Currently, there is no single diagnostic test for RLS. The disorder is diagnosed clinically by evaluating the patient's history and symptoms. Despite a clear description of clinical features, the condition is often misdiagnosed or underdiagnosed. In 1995, the International Restless Legs Syndrome Study Group identified four basic criteria for diagnosing RLS: (1) a desire to move the limbs, often associated with paresthesias or dysesthesias, (2) symptoms that are worse or present only during rest and are partially or temporarily relieved by activity, (3) motor restlessness, and (4) nocturnal worsening of symptoms. Although about 80 percent of those with RLS also experience PLMD, it is not necessary for a diagnosis of RLS. In more severe cases, patients may experience dyskinesia (uncontrolled, often continuous movements) while awake, and some experience symptoms in one or both of their arms as well as their legs. Most people with RLS have sleep disturbances, largely because of the limb discomfort and jerking. The result is excessive daytime sleepiness and fatigue. Despite these efforts to establish standard criteria, the clinical diagnosis of RLS is difficult to make. Physicians must rely largely on patients' descriptions of symptoms and information from their medical history, including past medical problems, family history, and current medications. Patients may be asked about frequency, duration, and intensity of symptoms as well as their tendency toward daytime sleep patterns and sleepiness, disturbance of sleep, or daytime function. If a patient's history is suggestive of RLS, laboratory tests may be performed to rule out other conditions and support the diagnosis of RLS. Blood tests to exclude anemia, decreased iron stores, diabetes, and renal dysfunction should be performed. Electromyography and nerve conduction studies may also be recommended to measure electrical activity in muscles and nerves, and Doppler sonography may be used to evaluate muscle activity in the legs. Such tests can document any accompanying damage or disease in nerves and nerve roots (such as peripheral neuropathy and radiculopathy) or other leg-related movement disorders. Negative results from tests may indicate that the diagnosis is RLS. In some cases, sleep studies such as polysomnography (a test that records the patient's brain waves, heartbeat, and breathing during an entire night) are undertaken to identify the presence of PLMD. The diagnosis is especially difficult with children because the physician relies heavily on the patient's explanations of symptoms, which, given the nature of the symptoms of RLS, can be difficult for a child to describe. The syndrome can sometimes be misdiagnosed as "growing pains" or attention deficit disorder. How is restless legs syndrome treated?Although movement brings relief to those with RLS, it is generally only temporary. However, RLS can be controlled by finding any possible underlying disorder. Often, treating the associated medical condition, such as peripheral neuropathy or diabetes, will alleviate many symptoms. For patients with idiopathic RLS, treatment is directed toward relieving symptoms. For those with mild to moderate symptoms, prevention is key, and many physicians suggest certain lifestyle changes and activities to reduce or eliminate symptoms. Decreased use of caffeine, alcohol, and tobacco may provide some relief. Physicians may suggest that certain individuals take supplements to correct deficiencies in iron, folate, and magnesium. Studies also have shown that maintaining a regular sleep pattern can reduce symptoms. Some individuals, finding that RLS symptoms are minimized in the early morning, change their sleep patterns. Others have found that a program of regular moderate exercise helps them sleep better; on the other hand, excessive exercise has been reported by some patients to aggravate RLS symptoms. Taking a hot bath, massaging the legs, or using a heating pad or ice pack can help relieve symptoms in some patients. Although many patients find some relief with such measures, rarely do these efforts completely eliminate symptoms Physicians also may suggest a variety of medications to treat RLS. Generally, physicians choose from dopaminergics, benzodiazepines (central nervous system depressants), opioids, and anticonvulsants. Dopaminergic agents, largely used to treat Parkinson's disease, have been shown to reduce RLS symptoms and PLMD and are considered the initial treatment of choice. Good short-term results of treatment with levodopa plus carbidopa have been reported, although most patients eventually will develop augmentation, meaning that symptoms are reduced at night but begin to develop earlier in the day than usual. Dopamine agonists such as pergolide mesylate, pramipexole, and ropinirole hydrochloride may be effective in some patients and are less likely to cause augmentation. In 2005, ropinirole became the only drug approved by the U.S. Food and Drug Administration specifically for the treatment of moderate to severe RLS. The drug was first approved in 1997 for patients with Parkinson’s disease. Benzodiazepines (such as clonazepam and diazepam) may be prescribed for patients who have mild or intermittent symptoms. These drugs help patients obtain a more restful sleep but they do not fully alleviate RLS symptoms and can cause daytime sleepiness. Because these depressants also may induce or aggravate sleep apnea in some cases, they should not be used in people with this condition. For more severe symptoms, opioids such as codeine, propoxyphene, or oxycodone may be prescribed for their ability to induce relaxation and diminish pain. Side effects include dizziness, nausea, vomiting, and the risk of addiction. Anticonvulsants such as carbamazepine and gabapentin are also useful for some patients, as they decrease the sensory disturbances (creeping and crawling sensations). Dizziness, fatigue, and sleepiness are among the possible side effects. Unfortunately, no one drug is effective for everyone with RLS. What may be helpful to one individual may actually worsen symptoms for another. In addition, medications taken regularly may lose their effect, making it necessary to change medications periodically. What is the prognosis of people with restless legs?RLS is generally a lifelong condition for which there is no cure. Symptoms may gradually worsen with age, though more slowly for those with the idiopathic form of RLS than for patients who also suffer from an associated medical condition. Nevertheless, current therapies can control the disorder, minimizing symptoms and increasing periods of restful sleep. In addition, some patients have remissions, periods in which symptoms decrease or disappear for days, weeks, or months, although symptoms usually eventually reappear. A diagnosis of RLS does not indicate the onset of another neurological disease. What research is being done?Within the Federal Government, the National Institute of Neurological Disorders and Stroke (NINDS), one of the National Institutes of Health, has primary responsibility for conducting and supporting research on RLS. The goal of this research is to increase scientific understanding of RLS, find improved methods of diagnosing and treating the syndrome, and discover ways to prevent it. NINDS-supported researchers are investigating the possible role of dopamine function in RLS. Dopamine is a chemical messenger responsible for transmitting signals between one area of the brain, the substantia nigra, and the next relay station of the brain, the corpus striatum, to produce smooth, purposeful muscle activity. Researchers suspect that impaired transmission of dopamine signals may play a role in RLS. Additional research should provide new information about how RLS occurs and may help investigators identify more successful treatment options. The NINDS sponsored a workshop on dopamine in 1999 to help plan a course for future research on disorders such as RLS and recommend ways to advance and encourage research in this field. Participants' recommendations for further research included the development of an animal model of RLS; additional genetic, epidemiologic, and pathophysiologic investigations of RLS; efforts to define genetic and non-genetic forms of RLS; establishment of a brain tissue bank to aid investigators; continuing investigations on dopamine and RLS; and studies of PLMD as it relates to RLS. Research on pallidotomy, a surgical procedure in which a portion of the brain called the globus pallidus is lesioned, may contribute to a greater understanding of the pathophysiology of RLS and may lead to a possible treatment. A recent study by NINDS-funded researchers showed that a patient with RLS and Parkinson's disease benefited from a pallidotomy and obtained relief from the limb discomfort caused by RLS. Additional research must be conducted to duplicate these results in other patients and to learn whether pallidotomy would be effective in RLS patients who do not also have Parkinson's disease. In other related research, NINDS scientists are conducting studies with patients to better understand the physiological mechanisms of PLMD associated with RLS. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 01-4847 |
|
Introduction
What is Epilepsy? What Causes Epilepsy? Genetic Factors Other Disorders Head Injury Prenatal Injury and Developmental Problems Poisoning What Are the Different Kinds of Seizures? Focal Seizures Generalized Seizures What Are the Different Kinds of Epilepsy? When Are Seizures Not Epilepsy? First Seizures Febrile Seizures Nonepileptic Events Eclampsia How is Epilepsy Diagnosed? EEG Monitoring Brain Scans Medical History Blood Tests Developmental, Neurological, and Behavioral Tests Can Epilepsy be Prevented? How can Epilepsy be Treated? Medications Tailoring the dosage of antiepileptic drugs Discontinuing medication Surgery Surgery to treat underlying conditions Surgery to remove a seizure focus Multiple subpial transection Corpus callosotomy Hemispherectomy and hemispherotomy Devices Diet Other Treatment Strategies How Does Epilepsy Affect Daily Life? Behavior and Emotions Driving and Recreation Education and Employment Pregnancy and Motherhood Are There Special Risks Associated With Epilepsy? Status Epilepticus Sudden Unexplained Death What Research Is Being Done on Epilepsy? How Can I Help Research on Epilepsy? What To Do If You See Someone Having a Seizure Conclusion Where can I get more information? Glossary IntroductionFew experiences match the drama of a convulsive seizure. A person having a severe seizure may cry out, fall to the floor unconscious, twitch or move uncontrollably, drool, or even lose bladder control. Within minutes, the attack is over, and the person regains consciousness but is exhausted and dazed. This is the image most people have when they hear the word epilepsy. However, this type of seizure -- a generalized tonic-clonic seizure -- is only one kind of epilepsy. There are many other kinds, each with a different set of symptoms. Epilepsy was one of the first brain disorders to be described. It was mentioned in ancient Babylon more than 3,000 years ago. The strange behavior caused by some seizures has contributed through the ages to many superstitions and prejudices. The word epilepsy is derived from the Greek word for "attack." People once thought that those with epilepsy were being visited by demons or gods. However, in 400 B.C., the early physician Hippocrates suggested that epilepsy was a disorder of the brain -- and we now know that he was right. What is Epilepsy?Epilepsy is a brain disorder in which clusters of nerve cells, or neurons, in the brain sometimes signal abnormally. Neurons normally generate electrochemical impulses that act on other neurons, glands, and muscles to produce human thoughts, feelings, and actions. In epilepsy, the normal pattern of neuronal activity becomes disturbed, causing strange sensations, emotions, and behavior, or sometimes convulsions , muscle spasms, and loss of consciousness. During a seizure, neurons may fire as many as 500 times a second, much faster than normal. In some people, this happens only occasionally; for others, it may happen up to hundreds of times a day. More than 2 million people in the United States -- about 1 in 100 -- have experienced an unprovoked seizure or been diagnosed with epilepsy. For about 80 percent of those diagnosed with epilepsy, seizures can be controlled with modern medicines and surgical techniques. However, about 25 to 30 percent of people with epilepsy will continue to experience seizures even with the best available treatment. Doctors call this situation intractable epilepsy. Having a seizure does not necessarily mean that a person has epilepsy. Only when a person has had two or more seizures is he or she considered to have epilepsy. Epilepsy is not contagious and is not caused by mental illness or mental retardation. Some people with mental retardation may experience seizures, but seizures do not necessarily mean the person has or will develop mental impairment. Many people with epilepsy have normal or above-average intelligence. Famous people who are known or rumored to have had epilepsy include the Russian writer Dostoyevsky, the philosopher Socrates, the military general Napoleon, and the inventor of dynamite, Alfred Nobel, who established the Nobel Prize. Several Olympic medalists and other athletes also have had epilepsy. Seizures sometimes do cause brain damage, particularly if they are severe. However, most seizures do not seem to have a detrimental effect on the brain. Any changes that do occur are usually subtle, and it is often unclear whether these changes are caused by the seizures themselves or by the underlying problem that caused the seizures. While epilepsy cannot currently be cured, for some people it does eventually go away. One study found that children with idiopathic epilepsy, or epilepsy with an unknown cause, had a 68 to 92 percent chance of becoming seizure-free by 20 years after their diagnosis. The odds of becoming seizure-free are not as good for adults or for children with severe epilepsy syndromes, but it is nonetheless possible that seizures may decrease or even stop over time. This is more likely if the epilepsy has been well-controlled by medication or if the person has had epilepsy surgery. What Causes Epilepsy?Epilepsy is a disorder with many possible causes. Anything that disturbs the normal pattern of neuron activity -- from illness to brain damage to abnormal brain development -- can lead to seizures. Epilepsy may develop because of an abnormality in brain wiring, an imbalance of nerve signaling chemicals called neurotransmitters, or some combination of these factors. Researchers believe that some people with epilepsy have an abnormally high level of excitatory neurotransmitters that increase neuronal activity, while others have an abnormally low level of inhibitory neurotransmitters that decrease neuronal activity in the brain. Either situation can result in too much neuronal activity and cause epilepsy. One of the most-studied neurotransmitters that plays a role in epilepsy is GABA, or gamma-aminobutyric acid, which is an inhibitory neurotransmitter. Research on GABA has led to drugs that alter the amount of this neurotransmitter in the brain or change how the brain responds to it. Researchers also are studying excitatory neurotransmitters such as glutamate. In some cases, the brain's attempts to repair itself after a head injury, stroke, or other problem may inadvertently generate abnormal nerve connections that lead to epilepsy. Abnormalities in brain wiring that occur during brain development also may disturb neuronal activity and lead to epilepsy. Research has shown that the cell membrane that surrounds each neuron plays an important role in epilepsy. Cell membranes are crucial for a neuron to generate electrical impulses. For this reason, researchers are studying details of the membrane structure, how molecules move in and out of membranes, and how the cell nourishes and repairs the membrane. A disruption in any of these processes may lead to epilepsy. Studies in animals have shown that, because the brain continually adapts to changes in stimuli, a small change in neuronal activity, if repeated, may eventually lead to full-blown epilepsy. Researchers are investigating whether this phenomenon, called kindling, may also occur in humans. In some cases, epilepsy may result from changes in non-neuronal brain cells called glia. These cells regulate concentrations of chemicals in the brain that can affect neuronal signaling. About half of all seizures have no known cause. However, in other cases, the seizures are clearly linked to infection, trauma, or other identifiable problems. Genetic FactorsResearch suggests that genetic abnormalities may be some of the most important factors contributing to epilepsy. Some types of epilepsy have been traced to an abnormality in a specific gene. Many other types of epilepsy tend to run in families, which suggests that genes influence epilepsy. Some researchers estimate that more than 500 genes could play a role in this disorder. However, it is increasingly clear that, for many forms of epilepsy, genetic abnormalities play only a partial role, perhaps by increasing a person's susceptibility to seizures that are triggered by an environmental factor. Several types of epilepsy have now been linked to defective genes for ion channels, the "gates" that control the flow of ions in and out of cells and regulate neuron signaling. Another gene, which is missing in people with progressive myoclonus epilepsy, codes for a protein called cystatin B. This protein regulates enzymes that break down other proteins. Another gene, which is altered in a severe form of epilepsy called LaFora's disease, has been linked to a gene that helps to break down carbohydrates. While abnormal genes sometimes cause epilepsy, they also may influence the disorder in subtler ways. For example, one study showed that many people with epilepsy have an abnormally active version of a gene that increases resistance to drugs. This may help explain why anticonvulsant drugs do not work for some people. Genes also may control other aspects of the body's response to medications and each person's susceptibility to seizures, or seizure threshold. Abnormalities in the genes that control neuronal migration -- a critical step in brain development -- can lead to areas of misplaced or abnormally formed neurons, or dysplasia, in the brain that can cause epilepsy. In some cases, genes may contribute to development of epilepsy even in people with no family history of the disorder. These people may have a newly developed abnormality, or mutation, in an epilepsy-related gene. Other DisordersIn many cases, epilepsy develops as a result of brain damage from other disorders. For example, brain tumors, alcoholism, and Alzheimer's disease frequently lead to epilepsy because they alter the normal workings of the brain. Strokes, heart attacks, and other conditions that deprive the brain of oxygen also can cause epilepsy in some cases. About 32 percent of all cases of newly developed epilepsy in elderly people appears to be due to cerebrovascular disease, which reduces the supply of oxygen to brain cells. Meningitis, AIDS, viral encephalitis, and other infectious diseases can lead to epilepsy, as can hydrocephalus -- a condition in which excess fluid builds up in the brain. Epilepsy also can result from intolerance to wheat gluten (also known as celiac disease), or from a parasitic infection of the brain called neurocysticercosis. Seizures may stop once these disorders are treated successfully. However, the odds of becoming seizure-free after the primary disorder is treated are uncertain and vary depending on the type of disorder, the brain region that is affected, and how much brain damage occurred prior to treatment. Epilepsy is associated with a variety of developmental and metabolic disorders, including cerebral palsy, neurofibromatosis, pyruvate dependency, tuberous sclerosis, Landau-Kleffner syndrome, and autism. Epilepsy is just one of a set of symptoms commonly found in people with these disorders. Head InjuryIn some cases, head injury can lead to seizures or epilepsy. Safety measures such as wearing seat belts in cars and using helmets when riding a motorcycle or playing competitive sports can protect people from epilepsy and other problems that result from head injury. Prenatal Injury and Developmental ProblemsThe developing brain is susceptible to many kinds of injury. Maternal infections, poor nutrition, and oxygen deficiencies are just some of the conditions that may take a toll on the brain of a developing baby. These conditions may lead to cerebral palsy, which often is associated with epilepsy, or they may cause epilepsy that is unrelated to any other disorders. About 20 percent of seizures in children are due to cerebral palsy or other neurological abnormalities. Abnormalities in genes that control development also may contribute to epilepsy. Advanced brain imaging has revealed that some cases of epilepsy that occur with no obvious cause may be associated with areas of dysplasia in the brain that probably develop before birth. PoisoningSeizures can result from exposure to lead, carbon monoxide, and many other poisons. They also can result from exposure to street drugs and from overdoses of antidepressants and other medications. Seizures are often triggered by factors such as lack of sleep, alcohol consumption, stress, or hormonal changes associated with the menstrual cycle. These seizure triggers do not cause epilepsy but can provoke first seizures or cause breakthrough seizures in people who otherwise experience good seizure control with their medication. Sleep deprivation in particular is a universal and powerful trigger of seizures. For this reason, people with epilepsy should make sure to get enough sleep and should try to stay on a regular sleep schedule as much as possible. For some people, light flashing at a certain speed or the flicker of a computer monitor can trigger a seizure; this problem is called photosensitive epilepsy. Smoking cigarettes also can trigger seizures. The nicotine in cigarettes acts on receptors for the excitatory neurotransmitter acetylcholine in the brain, which increases neuronal firing. Seizures are not triggered by sexual activity except in very rare instances. What Are the Different Kinds of Seizures?Doctors have described more than 30 different types of seizures. Seizures are divided into two major categories -- focal seizures and generalized seizures. However, there are many different types of seizures in each of these categories. Focal SeizuresFocal seizures, also called partial seizures, occur in just one part of the brain. About 60 percent of people with epilepsy have focal seizures. These seizures are frequently described by the area of the brain in which they originate. For example, someone might be diagnosed with focal frontal lobe seizures. In a simple focal seizure, the person will remain conscious but experience unusual feelings or sensations that can take many forms. The person may experience sudden and unexplainable feelings of joy, anger, sadness, or nausea. He or she also may hear, smell, taste, see, or feel things that are not real. In a complex focal seizure, the person has a change in or loss of consciousness. His or her consciousness may be altered, producing a dreamlike experience. People having a complex focal seizure may display strange, repetitious behaviors such as blinks, twitches, mouth movements, or even walking in a circle. These repetitious movements are called automatisms. More complicated actions, which may seem purposeful, can also occur involuntarily. Patients may also continue activities they started before the seizure began, such as washing dishes in a repetitive, unproductive fashion. These seizures usually last just a few seconds. Some people with focal seizures, especially complex focal seizures, may experience auras -- unusual sensations that warn of an impending seizure. These auras are actually simple focal seizures in which the person maintains consciousness. The symptoms an individual person has, and the progression of those symptoms, tend to be stereotyped, or similar every time. The symptoms of focal seizures can easily be confused with other disorders. For instance, the dreamlike perceptions associated with a complex focal seizure may be misdiagnosed as migraine headaches, which also may cause a dreamlike state. The strange behavior and sensations caused by focal seizures also can be istaken for symptoms of narcolepsy, fainting, or even mental illness. It may take many tests and careful monitoring by an experienced physician to tell the difference between epilepsy and other disorders. Generalized SeizuresGeneralized seizures are a result of abnormal neuronal activity on both sides of the brain. These seizures may cause loss of consciousness, falls, or massive muscle spasms. There are many kinds of generalized seizures. In absence seizures, the person may appear to be staring into space and/or have jerking or twitching muscles. These seizures are sometimes referred to as petit mal seizures, which is an older term. Tonic seizures cause stiffening of muscles of the body, generally those in the back, legs, and arms. Clonic seizures cause repeated jerking movements of muscles on both sides of the body. Myoclonic seizures cause jerks or twitches of the upper body, arms, or legs. Atonic seizures cause a loss of normal muscle tone. The affected person will fall down or may drop his or her head involuntarily. Tonic-clonic seizures cause a mixture of symptoms, including stiffening of the body and repeated jerks of the arms and/or legs as well as loss of consciousness. Tonic-clonic seizures are sometimes referred to by an older term: grand mal seizures. Not all seizures can be easily defined as either focal or generalized. Some people have seizures that begin as focal seizures but then spread to the entire brain. Other people may have both types of seizures but with no clear pattern. Society's lack of understanding about the many different types of seizures is one of the biggest problems for people with epilepsy. People who witness a non-convulsive seizure often find it difficult to understand that behavior which looks deliberate is not under the person's control. In some cases, this has led to the affected person being arrested or admitted to a psychiatric hospital. To combat these problems, people everywhere need to understand the many different types of seizures and how they may appear. What Are the Different Kinds of Epilepsy?Just as there are many different kinds of seizures, there are many different kinds of epilepsy. Doctors have identified hundreds of different epilepsy syndromes -- disorders characterized by a specific set of symptoms that include epilepsy. Some of these syndromes appear to be hereditary. For other syndromes, the cause is unknown. Epilepsy syndromes are frequently described by their symptoms or by where in the brain they originate. People should discuss the implications of their type of epilepsy with their doctors to understand the full range of symptoms, the possible treatments, and the prognosis. People with absence epilepsy have repeated absence seizures that cause momentary lapses of consciousness. These seizures almost always begin in childhood or adolescence, and they tend to run in families, suggesting that they may be at least partially due to a defective gene or genes. Some people with absence seizures have purposeless movements during their seizures, such as a jerking arm or rapidly blinking eyes. Others have no noticeable symptoms except for brief times when they are "out of it." Immediately after a seizure, the person can resume whatever he or she was doing. However, these seizures may occur so frequently that the person cannot concentrate in school or other situations. Childhood absence epilepsy usually stops when the child reaches puberty. Absence seizures usually have no lasting effect on intelligence or other brain functions. Temporal lobe epilepsy, or TLE, is the most common epilepsy syndrome with focal seizures. These seizures are often associated with auras. TLE often begins in childhood. Research has shown that repeated temporal lobe seizures can cause a brain structure called the hippocampus to shrink over time. The hippocampus is important for memory and learning. While it may take years of temporal lobe seizures for measurable hippocampal damage to occur, this finding underlines the need to treat TLE early and as effectively as possible. Neocortical epilepsy is characterized by seizures that originate from the brain's cortex, or outer layer. The seizures can be either focal or generalized. They may include strange sensations, visual hallucinations, emotional changes, muscle spasms, convulsions, and a variety of other symptoms, depending on where in the brain the seizures originate. There are many other types of epilepsy, each with its own characteristic set of symptoms. Many of these, including Lennox-Gastaut syndrome and Rasmussen's encephalitis, begin in childhood. Children with Lennox-Gastaut syndrome have severe epilepsy with several different types of seizures, including atonic seizures, which cause sudden falls and are also called drop attacks. This severe form of epilepsy can be very difficult to treat effectively. Rasmussen's encephalitis is a progressive type of epilepsy in which half of the brain shows continual inflammation. It sometimes is treated with a radical surgical procedure called hemispherectomy (see the section on Surgery). Some childhood epilepsy syndromes, such as childhood absence epilepsy, tend to go into remission or stop entirely during adolescence, whereas other syndromes such as juvenile myoclonic epilepsy and Lennox-Gastaut syndrome are usually present for life once they develop. Seizure syndromes do not always appear in childhood, however. Epilepsy syndromes that are easily treated, do not seem to impair cognitive functions or development, and usually stop spontaneously are often described as benign. Benign epilepsy syndromes include benign infantile encephalopathy and benign neonatal convulsions. Other syndromes, such as early myoclonic encephalopathy, include neurological and developmental problems. However, these problems may be caused by underlying neurodegenerative processes rather than by the seizures. Epilepsy syndromes in which the seizures and/or the person's cognitive abilities get worse over time are called progressive epilepsy. Several types of epilepsy begin in infancy. The most common type of infantile epilepsy is infantile spasms, clusters of seizures that usually begin before the age of 6 months. During these seizures the infant may bend and cry out. Anticonvulsant drugs often do not work for infantile spasms, but the seizures can be treated with ACTH (adrenocorticotropic hormone) or prednisone. When Are Seizures Not Epilepsy?While any seizure is cause for concern, having a seizure does not by itself mean a person has epilepsy. First seizures, febrile seizures, nonepileptic events, and eclampsia are examples of seizures that may not be associated with epilepsy. First SeizuresMany people have a single seizure at some point in their lives. Often these seizures occur in reaction to anesthesia or a strong drug, but they also may be unprovoked, meaning that they occur without any obvious triggering factor. Unless the person has suffered brain damage or there is a family history of epilepsy or other neurological abnormalities, these single seizures usually are not followed by additional seizures. One recent study that followed patients for an average of 8 years found that only 33 percent of people have a second seizure within 4 years after an initial seizure. People who did not have a second seizure within that time remained seizure-free for the rest of the study. For people who did have a second seizure, the risk of a third seizure was about 73 percent on average by the end of 4 years. When someone has experienced a first seizure, the doctor will usually order an electroencephalogram, or EEG, to determine what type of seizure the person may have had and if there are any detectable abnormalities in the person's brain waves. Thedoctor also may order brain scans to identify abnormalities that may be visible in the brain. These tests may help the doctor decide whether or not to treat the person with antiepileptic drugs. In some cases, drug treatment after the first seizure may help prevent future seizures and epilepsy. However, the drugs also can cause detrimental side effects, so doctors prescribe them only when they feel the benefits outweigh the risks. Evidence suggests that it may be beneficial to begin anticonvulsant medication once a person has had a second seizure, as the chance of future seizures increases significantly after this occurs. Febrile SeizuresSometimes a child will have a seizure during the course of an illness with a high fever. These seizures are called febrile seizures (febrile is derived from the Latin word for "fever") and can be very alarming to the parents and other caregivers. In the past, doctors usually prescribed a course of anticonvulsant drugs following a febrile seizure in the hope of preventing epilepsy. However, most children who have a febrile seizure do not develop epilepsy, and long-term use of anticonvulsant drugs in children may damage the developing brain or cause other detrimental side effects. Experts at a 1980 consensus conference coordinated by the National Institutes of Health concluded that preventive treatment after a febrile seizure is generally not warranted unless certain other conditions are present: a family history of epilepsy, signs of nervous system impairment prior to the seizure, or a relatively prolonged or complicated seizure. The risk of subsequent non-febrile seizures is only 2 to 3 percent unless one of these factors is present. Researchers have now identified several different genes that influence the risk of febrile seizures in certain families. Studying these genes may lead to new understanding of how febrile seizures occur and perhaps point to ways of preventing them. Nonepileptic EventsSometimes people appear to have seizures, even though their brains show no seizure activity. This type of phenomenon has various names, including nonepileptic events and pseudoseizures. Both of these terms essentially mean something that looks like a seizure but isn't one. Nonepileptic events that are psychological in origin may be referred to as psychogenic seizures. Psychogenic seizures may indicate dependence, a need for attention, avoidance of stressful situations, or specific psychiatric conditions. Some people with epilepsy have psychogenic seizures in addition to their epileptic seizures. Other people who have psychogenic seizures do not have epilepsy at all. Psychogenic seizures cannot be treated in the same way as epileptic seizures. Instead, they are often treated by mental health specialists. Other nonepileptic events may be caused by narcolepsy, Tourette syndrome, cardiac arrythmia, and other medical conditions with symptoms that resemble seizures. Because symptoms of these disorders can look very much like epileptic seizures, they are often mistaken for epilepsy. Distinguishing between true epileptic seizures and nonepileptic events can be very difficult and requires a thorough medical assessment, careful monitoring, and knowledgeable health professionals. Improvements in brain scanning and monitoring technology may improve diagnosis of nonepileptic events in the future. EclampsiaEclampsia is a life-threatening condition that can develop in pregnant women. Its symptoms include sudden elevations of blood pressure and seizures. Pregnant women who develop unexpected seizures should be rushed to a hospital immediately. Eclampsia can be treated in a hospital setting and usually does not result in additional seizures or epilepsy once the pregnancy is over. How is Epilepsy Diagnosed?Doctors have developed a number of different tests to determine whether a person has epilepsy and, if so, what kind of seizures the person has. In some cases, people may have symptoms that look very much like a seizure but in fact are nonepileptic events caused by other disorders. Even doctors may not be able to tell the difference between these disorders and epilepsy without close observation and intensive testing. EEG MonitoringAn EEG records brain waves detected by electrodes placed on the scalp. This is the most common diagnostic test for epilepsy and can detect abnormalities in the brain's electrical activity. People with epilepsy frequently have changes in their normal pattern of brain waves, even when they are not experiencing a seizure. While this type of test can be very useful in diagnosing epilepsy, it is not foolproof. Some people continue to show normal brain wave patterns even after they have experienced a seizure. In other cases, the unusual brain waves are generated deep in the brain where the EEG is unable to detect them. Many people who do not have epilepsy also show some unusual brain activity on an EEG. Whenever possible, an EEG should be performed within 24 hours of a patient's first seizure. Ideally, EEGs should be performed while the patient is sleeping as well as when he or she is awake, because brain activity during sleep is often quite different than at other times. Video monitoring is often used in conjunction with EEG to determine the nature of a person's seizures. It also can be used in some cases to rule out other disorders such as cardiac arrythmia or narcolepsy that may look like epilepsy. Brain ScansOne of the most important ways of diagnosing epilepsy is through the use of brain scans. The most commonly used brain scans include CT (computed tomography), PET (positron emission tomography) and MRI (magnetic resonance imaging). CT and MRI scans reveal the structure of the brain, which can be useful for identifying brain tumors, cysts, and other structural abnormalities. PET and an adapted kind of MRI called functional MRI (fMRI) can be used to monitor the brain's activity and detect abnormalities in how it works. SPECT (single photon emission computed tomography) is a relatively new kind of brain scan that is sometimes used to locate seizure foci in the brain. In some cases, doctors may use an experimental type of brain scan called a magnetoencephalogram, or MEG. MEG detects the magnetic signals generated by neurons to allow doctors to monitor brain activity at different points in the brain over time, revealing different brain functions. While MEG is similar in concept to EEG, it does not require electrodes and it can detect signals from deeper in the brain than an EEG. Doctors also are experimenting with brain scans called magnetic resonance spectroscopy (MRS) that can detect abnormalities in the brain's biochemical processes, and with near-infrared spectroscopy, a technique that can detect oxygen levels in brain tissue. Medical HistoryTaking a detailed medical history, including symptoms and duration of the seizures, is still one of the best methods available to determine if a person has epilepsy and what kind of seizures he or she has. The doctor will ask questions about the seizures and any past illnesses or other symptoms a person may have had. Since people who have suffered a seizure often do not remember what happened, caregivers' accounts of the seizure are vital to this evaluation. Blood TestsDoctors often take blood samples for testing, particularly when they are examining a child. These blood samples are often screened for metabolic or genetic disorders that may be associated with the seizures. They also may be used to check for underlying problems such as infections, lead poisoning, anemia, and diabetes that may be causing or triggering the seizures. Developmental, Neurological, and Behavioral TestsDoctors often use tests devised to measure motor abilities, behavior, and intellectual capacity as a way to determine how the epilepsy is affecting that person. These tests also can provide clues about what kind of epilepsy the person has. Can Epilepsy be Prevented?Many cases of epilepsy can be prevented by wearing seatbelts and bicycle helmets, putting children in car seats, and other measures that prevent head injury and other trauma. Prescribing medication after first or second seizures or febrile seizures also may help prevent epilepsy in some cases. Good prenatal care, including treatment of high blood pressure and infections during pregnancy, can prevent brain damage in the developing baby that may lead to epilepsy and other neurological problems later. Treating cardiovascular disease, high blood pressure, infections, and other disorders that can affect the brain during adulthood and aging also may prevent many cases of epilepsy. Finally, identifying the genes for many neurological disorders can provide opportunities for genetic screening and prenatal diagnosis that may ultimately prevent many cases of epilepsy. How can Epilepsy be Treated?Accurate diagnosis of the type of epilepsy a person has is crucial for finding an effective treatment. There are many different ways to treat epilepsy. Currently available treatments can control seizures at least some of the time in about 80 percent of people with epilepsy. However, another 20 percent -- about 600,000 people with epilepsy in the United States -- have intractable seizures, and another 400,000 feel they get inadequate relief from available treatments. These statistics make it clear that improved treatments are desperately needed. Doctors who treat epilepsy come from many different fields of medicine. They include neurologists, pediatricians, pediatric neurologists, internists, and family physicians, as well as neurosurgeons and doctors called epileptologists who specialize in treating epilepsy. People who need specialized or intensive care for epilepsy may be treated at large medical centers and neurology clinics at hospitals or by neurologists in private practice. Many epilepsy treatment centers are associated with university hospitals that perform research in addition to providing medical care. Once epilepsy is diagnosed, it is important to begin treatment as soon as possible. Research suggests that medication and other treatments may be less successful in treating epilepsy once seizures and their consequences become established. MedicationsBy far the most common approach to treating epilepsy is to prescribe antiepileptic drugs. The first effective antiepileptic drugs were bromides, introduced by an English physician named Sir Charles Locock in 1857. He noticed that bromides had a sedative effect and seemed to reduce seizures in some patients. More than 20 different antiepileptic drugs are now on the market, all with different benefits and side effects. The choice of which drug to prescribe, and at what dosage, depends on many different factors, including the type of seizures a person has, the person’s lifestyle and age, how frequently the seizures occur, and, for a woman, the likelihood that she will become pregnant. People with epilepsy should follow their doctor’s advice and share any concerns they may have regarding their medication. Doctors seeing a patient with newly developed epilepsy often prescribe carbamazepine, valproate, lamotrigine, oxcarbazepine, or phenytoin first, unless the epilepsy is a type that is known to require a different kind of treatment. For absence seizures, ethosuximide is often the primary treatment. Other commonly prescribed drugs include clonazepam, phenobarbital, and primidone. Some relatively new epilepsy drugs include tiagabine, gabapentin, topiramate, levetiracetam, and felbamate. Other drugs are used in combination with one of the standard drugs or for intractable seizures that do not respond to other medications. A few drugs, such as fosphenytoin, are approved for use only in hospital settings to treat specific problems such as status epilepticus (see section, “Are There Special Risks Associated With Epilepsy?” ). For people with stereotyped recurrent severe seizures that can be easily recognized by the person’s family, the drug diazepam is now available as a gel that can be administered rectally by a family member. This method of drug delivery may be able to stop prolonged or repeated seizures before they develop into status epilepticus. For most people with epilepsy, seizures can be controlled with just one drug at the optimal dosage. Combining medications usually amplifies side effects such as fatigue and decreased appetite, so doctors usually prescribe monotherapy, or the use of just one drug, whenever possible. Combinations of drugs are sometimes prescribed if monotherapy fails to effectively control a patient’s seizures. The number of times a person needs to take medication each day is usually determined by the drug’s half-life, or the time it takes for half the drug dose to be metabolized or broken down into other substances in the body. Some drugs, such as phenytoin and phenobarbital, only need to be taken once a day, while others such as valproate must be taken two or three times a day. Most side effects of antiepileptic drugs are relatively minor, such as fatigue, dizziness, or weight gain. However, severe and life-threatening side effects such as allergic reactions can occur. Epilepsy medication also may predispose people to developing depression or psychoses. People with epilepsy should consult a doctor immediately if they develop any kind of rash while on medication, or if they find themselves depressed or otherwise unable to think in a rational manner. Other danger signs that should be discussed with a doctor immediately are extreme fatigue, staggering or other movement problems, and slurring of words. People with epilepsy should be aware that their epilepsy medication can interact with many other drugs in potentially harmful ways. For this reason, people with epilepsy should always tell doctors who treat them which medications they are taking. Women also should know that some antiepileptic drugs can interfere with the effectiveness of oral contraceptives, and they should discuss this possibility with their doctors. Since people can become more sensitive to medications as they age, they may need to have their blood levels of medication checked occasionally to see if the dose needs to be adjusted. The effects of a particular medication also sometimes wear off over time, leading to an increase in seizures if the dose is not adjusted. People should know that some citrus fruit, in particular grapefruit juice, may interfere with breakdown of many drugs. This can cause too much of the drug to build up in their bodies, often worsening the side effects. People taking epilepsy medication should be sure to check with their doctor and/or seek a second medical opinion if their medication does not appear to be working or if it causes unexpected side effects. Tailoring the dosage of antiepileptic drugsWhen a person starts a new epilepsy drug, it is important to tailor the dosage to achieve the best results. People's bodies react to medications in very different and sometimes unpredictable ways, so it may take some time to find the right drug at the right dose to provide optimal control of seizures while minimizing side effects. A drug that has no effect or very bad side effects at one dose may work very well at another dose. Doctors will usually prescribe a low dose of the new drug initially and monitor blood levels of the drug to determine when the best possible dose has been reached. Generic versions are available for many antiepileptic drugs. The chemicals in generic drugs are exactly the same as in the brand-name drugs, but they may be absorbed or processed differently in the body because of the way they are prepared. Therefore, patients should always check with their doctors before switching to a generic version of their medication. Discontinuing medicationSome doctors will advise people with epilepsy to discontinue their antiepileptic drugs after 2 years have passed without a seizure. Others feel it is better to wait for 4 to 5 years. Discontinuing medication should always be done with a doctor's advice and supervision. It is very important to continue taking epilepsy medication for as long as the doctor prescribes it. People also should ask the doctor or pharmacist ahead of time what they should do if they miss a dose. Discontinuing medication without a doctor's advice is one of the major reasons people who have been seizure-free begin having new seizures. Seizures that result from suddenly stopping medication can be very serious and can lead to status epilepticus. Furthermore, there is some evidence that uncontrolled seizures trigger changes in neurons that can make it more difficult to treat the seizures in the future. The chance that a person will eventually be able to discontinue medication varies depending on the person's age and his or her type of epilepsy. More than half of children who go into remission with medication can eventually stop their medication without having new seizures. One study showed that 68 percent of adults who had been seizure-free for 2 years before stopping medication were able to do so without having more seizures and 75 percent could successfully discontinue medication if they had been seizure-free for 3 years. However, the odds of successfully stopping medication are not as good for people with a family history of epilepsy, those who need multiple medications, those with focal seizures, and those who continue to have abnormal EEG results while on medication. SurgeryWhen seizures cannot be adequately controlled by medications, doctors may recommend that the person be evaluated for surgery. Surgery for epilepsy is performed by teams of doctors at medical centers. To decide if a person may benefit from surgery, doctors consider the type or types of seizures he or she has. They also take into account the brain region involved and how important that region is for everyday behavior. Surgeons usually avoid operating in areas of the brain that are necessary for speech, language, hearing, or other important abilities. Doctors may perform tests such as a Wada test (administration of the drug amobarbitol into the carotid artery) to find areas of the brain that control speech and memory. They often monitor the patient intensively prior to surgery in order to pinpoint the exact location in the brain where seizures begin. They also may use implanted electrodes to record brain activity from the surface of the brain. This yields better information than an external EEG. A 1990 National Institutes of Health consensus conference on surgery for epilepsy concluded that there are three broad categories of epilepsy that can be treated successfully with surgery. These include focal seizures, seizures that begin as focal seizures before spreading to the rest of the brain, and unilateral multifocal epilepsy with infantile hemiplegia (such as Rasmussen's encephalitis). Doctors generally recommend surgery only after patients have tried two or three different medications without success, or if there is an identifiable brain lesion--a damaged or dysfunctional area--believed to cause the seizures. A study published in 2000 compared surgery to an additional year of treatment with antiepileptic drugs in people with longstanding temporal lobe epilepsy. The results showed that 64 percent of patients receiving surgery became seizure-free, compared to 8 percent of those who continued with medication only. Because of this study and other evidence, the American Academy of Neurology (AAN) now recommends surgery for TLE when antiepileptic drugs are not effective. However, the study and the AAN guidelines do not provide guidance on how long seizures should occur, how severe they should be, or how many drugs should be tried before surgery is considered. A nationwide study is now underway to determine how soon surgery for TLE should be performed. If a person is considered a good candidate for surgery and has seizures that cannot be controlled with available medication, experts generally agree that surgery should be performed as early as possible. It can be difficult for a person who has had years of seizures to fully re-adapt to a seizure-free life if the surgery is successful. The person may never have had an opportunity to develop independence, and he or she may have had difficulties with school and work that could have been avoided with earlier treatment. Surgery should always be performed with support from rehabilitation specialists and counselors who can help the person deal with the many psychological, social, and employment issues he or she may face. While surgery can significantly reduce or even halt seizures for some people, it is important to remember that any kind of surgery carries some amount of risk (usually small). Surgery for epilepsy does not always successfully reduce seizures and it can result in cognitive or personality changes, even in people who are excellent candidates for surgery. Patients should ask their surgeon about his or her experience, success rates, and complication rates with the procedure they are considering. Even when surgery completely ends a person's seizures, it is important to continue taking seizure medication for some time to give the brain time to re-adapt. Doctors generally recommend medication for 2 years after a successful operation to avoid new seizures. Surgery to treat underlying conditionsIn cases where seizures are caused by a brain tumor, hydrocephalus, or other conditions that can be treated with surgery, doctors may operate to treat these underlying conditions. In many cases, once the underlying condition is successfully treated, a person's seizures will disappear as well. Surgery to remove a seizure focusThe most common type of surgery for epilepsy is removal of a seizure focus, or small area of the brain where seizures originate. This type of surgery, which doctors may refer to as a lobectomy or lesionectomy, is appropriate only for focal seizures that originate in just one area of the brain. In general, people have a better chance of becoming seizure-free after surgery if they have a small, well-defined seizure focus. Lobectomies have a 55-70 percent success rate when the type of epilepsy and the seizure focus is well-defined. The most common type of lobectomy is a temporal lobe resection, which is performed for people with temporal lobe epilepsy. Temporal lobe resection leads to a significant reduction or complete cessation of seizures about 70 - 90 percent of the time. Multiple subpial transectionWhen seizures originate in part of the brain that cannot be removed, surgeons may perform a procedure called a multiple subpial transection. In this type of operation, which has been commonly performed since 1989, surgeons make a series of cuts that are designed to prevent seizures from spreading into other parts of the brain while leaving the person's normal abilities intact. About 70 percent of patients who undergo a multiple subpial transection have satisfactory improvement in seizure control. Corpus callosotomyCorpus callosotomy, or severing the network of neural connections between the right and left halves, or hemispheres, of the brain, is done primarily in children with severe seizures that start in one half of the brain and spread to the other side. Corpus callosotomy can end drop attacks and other generalized seizures. However, the procedure does not stop seizures in the side of the brain where they originate, and these focal seizures may even increase after surgery. Hemispherectomy and hemispherotomyThese procedures remove half of the brain's cortex, or outer layer. They are used predominantly in children who have seizures that do not respond to medication because of damage that involves only half the brain, as occurs with conditions such as Rasmussen's encephalitis, Sturge-Weber syndrome, and hemimegencephaly. While this type of surgery is very radical and is performed only as a last resort, children often recover very well from the procedure, and their seizures usually cease altogether. With intense rehabilitation, they often recover nearly normal abilities. Since the chance of a full recovery is best in young children, hemispherectomy should be performed as early in a child's life as possible. It is rarely performed in children older than 13. DevicesThe vagus nerve stimulator was approved by the U.S. Food and Drug Administration (FDA) in 1997 for use in people with seizures that are not well-controlled by medication. The vagus nerve stimulator is a battery-powered device that is surgically implanted under the skin of the chest, much like a pacemaker, and is attached to the vagus nerve in the lower neck. This device delivers short bursts of electrical energy to the brain via the vagus nerve. On average, this stimulation reduces seizures by about 20 - 40 percent. Patients usually cannot stop taking epilepsy medication because of the stimulator, but they often experience fewer seizures and they may be able to reduce the dose of their medication. Side effects of the vagus nerve stimulator are generally mild but may include hoarseness, ear pain, a sore throat, or nausea. Adjusting the amount of stimulation can usually eliminate most side effects, although the hoarseness typically persists. The batteries in the vagus nerve stimulator need to be replaced about once every 5 years; this requires a minor operation that can usually be performed as an outpatient procedure. Several new devices may become available for epilepsy in the future. Researchers are studying whether transcranial magnetic stimulation (TMS), a procedure which uses a strong magnet held outside the head to influence brain activity, may reduce seizures. They also hope to develop implantable devices that can deliver drugs to specific parts of the brain. DietStudies have shown that, in some cases, children may experience fewer seizures if they maintain a strict diet rich in fats and low in carbohydrates. This unusual diet, called the ketogenic diet, causes the body to break down fats instead of carbohydrates to survive. This condition is called ketosis. One study of 150 children whose seizures were poorly controlled by medication found that about one-fourth of the children had a 90 percent or better decrease in seizures with the ketogenic diet, and another half of the group had a 50 percent or better decrease in their seizures. Moreover, some children can discontinue the ketogenic diet after several years and remain seizure-free. The ketogenic diet is not easy to maintain, as it requires strict adherence to an unusual and limited range of foods. Possible side effects include retarded growth due to nutritional deficiency and a buildup of uric acid in the blood, which can lead to kidney stones. People who try the ketogenic diet should seek the guidance of a dietician to ensure that it does not lead to serious nutritional deficiency. Researchers are not sure how ketosis inhibits seizures. One study showed that a byproduct of ketosis called beta-hydroxybutyrate (BHB) inhibits seizures in animals. If BHB also works in humans, researchers may eventually be able to develop drugs that mimic the seizure-inhibiting effects of the ketogenic diet. Other Treatment StrategiesResearchers are studying whether biofeedback -- a strategy in which individuals learn to control their own brain waves -- may be useful in controlling seizures. However, this type of therapy is controversial and most studies have shown discouraging results. Taking large doses of vitamins generally does not help a person's seizures and may even be harmful in some cases. But a good diet and some vitamin supplements, particularly folic acid, may help reduce some birth defects and medication-related nutritional deficiencies. Use of non-vitamin supplements such as melatonin is controversial and can be risky. One study showed that melatonin may reduce seizures in some children, while another found that the risk of seizures increased measurably with melatonin. Most non-vitamin supplements such as those found in health food stores are not regulated by the FDA, so their true effects and their interactions with other drugs are largely unknown. How Does Epilepsy Affect Daily Life?Most people with epilepsy lead outwardly normal lives. Approximately 80 percent can be significantly helped by modern therapies, and some may go months or years between seizures. However, the condition can and does affect daily life for people with epilepsy, their family, and their friends. People with severe seizures that resist treatment have, on average, a shorter life expectancy and an increased risk of cognitive impairment, particularly if the seizures developed in early childhood. These impairments may be related to the underlying conditions tha cause epilepsy or to epilepsy treatment rather than the epilepsy itself. Behavior and EmotionsIt is not uncommon for people with epilepsy, especially children, to develop behavioral and emotional problems. Sometimes these problems are caused by embarrassment or frustration associated with epilepsy. Other problems may result from bullying, teasing, or avoidance in school and other social settings. In children, these problems can be minimized if parents encourage a positive outlook and independence, do not reward negative behavior with unusual amounts of attention, and try to stay attuned to their child's needs and feelings. Families must learn to accept and live with the seizures without blaming or resenting the affected person. Counseling services can help families cope with epilepsy in a positive manner. Epilepsy support groups also can help by providing a way for people with epilepsy and their family members to share their experiences, frustrations, and tips for coping with the disorder. People with epilepsy have an increased risk of poor self-esteem, depression, and suicide. These problems may be a reaction to a lack of understanding or discomfort about epilepsy that may result in cruelty or avoidance by other people. Many people with epilepsy also live with an ever-present fear that they will have another seizure. Driving and RecreationFor many people with epilepsy, the risk of seizures restricts their independence, in particular the ability to drive. Most states and the District of Columbia will not issue a driver's license to someone with epilepsy unless the person can document that they have gone a specific amount of time without a seizure (the waiting period varies from a few months to several years). Some states make exceptions for this policy when seizures don't impair consciousness, occur only during sleep, or have long auras or other warning signs that allow the person to avoid driving when a seizure is likely to occur. Studies show that the risk of having a seizure-related accident decreases as the length of time since the last seizure increases. One study found that the risk of having a seizure-related motor vehicle accident is 93 percent less in people who wait at least 1 year after their last seizure before driving, compared to people who wait for shorter intervals. The risk of seizures also restricts people's recreational choices. For instance, people with epilepsy should not participate in sports such as skydiving or motor racing where a moment's inattention could lead to injury. Other activities, such as swimming and sailing, should be done only with precautions and/or supervision. However, jogging, football, and many other sports are reasonably safe for a person with epilepsy. Studies to date have not shown any increase in seizures due to sports, although these studies have not focused on any activity in particular. There is some evidence that regular exercise may even improve seizure control in some people. Sports are often such a positive factor in life that it is best for the person to participate, although the person with epilepsy and the coach or other leader should take appropriate safety precautions. It is important to take steps to avoid potential sports-related problems such as dehydration, overexertion, and hypoglycemia, as these problems can increase the risk of seizures. Education and EmploymentBy law, people with epilepsy or other handicaps in the United States cannot be denied employment or access to any educational, recreational, or other activity because of their seizures. However, one survey showed that only about 56 percent of people with epilepsy finish high school and about 15 percent finish college -- rates much lower than those for the general population. The same survey found that about 25 percent of working-age people with epilepsy are unemployed. These numbers indicate that significant barriers still exist for people with epilepsy in school and work. Restrictions on driving limit the employment opportunities for many people with epilepsy, and many find it difficult to face the misunderstandings and social pressures they encounter in public situations. Antiepileptic drugs also may cause side effects that interfere with concentration and memory. Children with epilepsy may need extra time to complete schoolwork, and they sometimes may need to have instructions or other information repeated for them. Teachers should be told what to do if a child in their classroom has a seizure, and parents should work with the school system to find reasonable ways to accommodate any special needs their child may have. Pregnancy and MotherhoodWomen with epilepsy are often concerned about whether they can become pregnant and have a healthy child. This is usually possible. While some seizure medications and some types of epilepsy may reduce a person's interest in sexual activity, most people with epilepsy can become pregnant. Moreover, women with epilepsy have a 90 percent or better chance of having a normal, healthy baby, and the risk of birth defects is only about 4 to 6 percent. The risk that children of parents with epilepsy will develop epilepsy themselves is only about 5 percent unless the parent has a clearly hereditary form of the disorder. Parents who are worried that their epilepsy may be hereditary may wish to consult a genetic counselor to determine what the risk might be. Amniocentesis and high-level ultrasound can be performed during pregnancy to ensure that the baby is developing normally, and a procedure called a maternal serum alpha-fetoprotein test can be used for prenatal diagnosis of many conditions if a problem is suspected. There are several precautions women can take before and during pregnancy to reduce the risks associated with pregnancy and delivery. Women who are thinking about becoming pregnant should talk with their doctors to learn any special risks associated with their epilepsy and the medications they may be taking. Some seizure medications, particularly valproate, trimethidone, and phenytoin, are known to increase the risk of having a child with birth defects such as cleft palate, heart problems, or finger and toe defects. For this reason, a woman's doctor may advise switching to other medications during pregnancy. Whenever possible, a woman should allow her doctor enough time to properly change medications, including phasing in the new medications and checking to determine when blood levels are stabilized, before she tries to become pregnant. Women should also begin prenatal vitamin supplements -- especially with folic acid, which may reduce the risk of some birth defects -- well before pregnancy. Women who discover that they are pregnant but have not already spoken with their doctor about ways to reduce the risks should do so as soon as possible. However, they should continue taking seizure medication as prescribed until that time to avoid preventable seizures. Seizures during pregnancy can harm the developing baby or lead to miscarriage, particularly if the seizures are severe. Nevertheless, many women who have seizures during pregnancy have normal, healthy babies. Women with epilepsy sometimes experience a change in their seizure frequency during pregnancy, even if they do not change medications. About 25 to 40 percent of women have an increase in their seizure frequency while they are pregnant, while other women may have fewer seizures during pregnancy. The frequency of seizures during pregnancy may be influenced by a variety of factors, including the woman's increased blood volume during pregnancy, which can dilute the effect of medication. Women should have their blood levels of seizure medications monitored closely during and after pregnancy, and the medication dosage should be adjusted accordingly. Pregnant women with epilepsy should take prenatal vitamins and get plenty of sleep to avoid seizures caused by sleep deprivation. They also should take vitamin K supplements after 34 weeks of pregnancy to reduce the risk of a blood-clotting disorder in infants called neonatal coagulopathy that can result from fetal exposure to epilepsy medications. Finally, they should get good prenatal care, avoid tobacco, caffeine, alcohol, and illegal drugs, and try to avoid stress. Labor and delivery usually proceed normally for women with epilepsy, although there is a slightly increased risk of hemorrhage, eclampsia, premature labor, and cesarean section. Doctors can administer antiepileptic drugs intravenously and monitor blood levels of anticonvulsant medication during labor to reduce the risk that the labor will trigger a seizure. Babies sometimes have symptoms of withdrawal from the mother's seizure medication after they are born, but these problems wear off in a few weeks or months and usually do not cause serious or long-term effects. A mother's blood levels of anticonvulsant medication should be checked frequently after delivery as medication often needs to be decreased. Epilepsy medications need not influence a woman's decision about breast-feeding her baby. Only minor amounts of epilepsy medications are secreted in breast milk, usually not enough to harm the baby and much less than the baby was exposed to in the womb. On rare occasions, the baby may become excessively drowsy or feed poorly, and these problems should be closely monitored. However, experts believe the benefits of breast-feeding outweigh the risks except in rare circumstances. To increase doctors' understanding of how different epilepsy medications affect pregnancy and the chances of having a healthy baby, Massachusetts General Hospital has begun a nationwide registry for women who take antiepileptic drugs while pregnant. Women who enroll in this program are given educational materials on pre-conception planning and perinatal care and are asked to provide information about the health of their children (this information is kept confidential). Women and physicians can contact this registry by calling 1-888-233-2334 or 617-726-1742 (fax: 617-724-8307). Women with epilepsy should be aware that some epilepsy medications can interfere with the effectiveness of oral contraceptives. Women who wish to use oral contraceptives to prevent pregnancy should discuss this with their doctors, who may be able to prescribe a different kind of antiepileptic medication or suggest other ways of avoiding an unplanned pregnancy. Are There Special Risks Associated With Epilepsy?Although most people with epilepsy lead full, active lives, they are at special risk for two life-threatening conditions: status epilepticus and sudden unexplained death. Status EpilepticusStatus epilepticus is a potentially life-threatening condition in which a person either has an abnormally prolonged seizure or does not fully regain consciousness between seizures. Although there is no strict definition for the time at which a seizure turns into status epilepticus, most people agree that any seizure lasting longer than 5 minutes should, for practical purposes, be treated as though it was status epilepticus. Status epilepticus affects about 195,000 people each year in the United States and results in about 42,000 deaths. While people with epilepsy are at an increased risk for status epilepticus, about 60 percent of people who develop this condition have no previous seizure history. These cases often result from tumors, trauma, or other problems that affect the brain and may themselves be life-threatening. While most seizures do not require emergency medical treatment, someone with a prolonged seizure lasting more than 5 minutes may be in status epilepticus and should be taken to an emergency room immediately. It is important to treat a person with status epilepticus as soon as possible. One study showed that 80 percent of people in status epilepticus who received medication within 30 minutes of seizure onset eventually stopped having seizures, whereas only 40 percent recovered if 2 hours had passed before they received medication. Doctors in a hospital setting can treat status epilepticus with several different drugs and can undertake emergency life-saving measures, such as administering oxygen, if necessary. People in status epilepticus do not always have severe convulsive seizures. Instead, they may have repeated or prolonged nonconvulsive seizures. This type of status epilepticus may appear as a sustained episode of confusion or agitation in someone who does not ordinarily have that kind of mental impairment. While this type of episode may not seem as severe as convulsive status epilepticus, it should still be treated as an emergency. Sudden Unexplained DeathFor reasons that are poorly understood, people with epilepsy have an increased risk of dying suddenly for no discernible reason. This condition, called sudden unexplained death, can occur in people without epilepsy, but epilepsy increases the risk about two-fold. Researchers are still unsure why sudden unexplained death occurs. One study suggested that use of more than two anticonvulsant drugs may be a risk factor. However, it is not clear whether the use of multiple drugs causes the sudden death, or whether people who use multiple anticonvulsants have a greater risk of death because they have more severe types of epilepsy. What Research Is Being Done on Epilepsy?While research has led to many advances in understanding and treating epilepsy, there are many unanswered questions about how and why seizures develop, how they can best be treated or prevented, and how they influence other brain activity and brain development. Researchers, many of whom are supported by the National Institute of Neurological Disorders and Stroke (NINDS), are studying all of these questions. They also are working to identify and test new drugs and other treatments for epilepsy and to learn how those treatments affect brain activity and development. The NINDS's Anticonvulsant Screening Program (ASP) studies potential new therapies with the goal of enhancing treatment for patients with epilepsy. Since it began in 1975, more than 390 public-private partnerships have been created. These partnerships have resulted in state-of-the-art evaluations of more than 25,000 compounds for their potential as antiepileptic drugs. This government-sponsored effort has contributed to the development of five drugs that are now approved for use in the United States. It has also aided in the discovery and profiling of six new compounds currently in various stages of clinical development. Besides testing for safer, more efficacious therapies, the Program is developing and validating new models that may one day find therapies that intervene in the disease process itself as well as models of resistant or refractory epilepsy. Scientists continue to study how excitatory and inhibitory neurotransmitters interact with brain cells to control nerve firing. They can apply different chemicals to cultures of neurons in laboratory dishes to study how those chemicals influence neuronal activity. They also are studying how glia and other non-neuronal cells in the brain contribute to seizures. This research may lead to new drugs and other new ways of treating seizures. Researchers also are working to identify genes that may influence epilepsy in some way. Identifying these genes can reveal the underlying chemical processes that influence epilepsy and point to new ways of preventing or treating this disorder. Researchers also can study rats and mice that have missing or abnormal copies of certain genes to determine how these genes affect normal brain development and resistance to damage from disease and other environmental factors. In the future, researchers may be able to use panels of gene fragments, called "gene chips," to determine each person's genetic makeup. This information may allow doctors to prevent epilepsy or to predict which treatments will be most beneficial. Doctors are now experimenting with several new types of therapies for epilepsy. In one preliminary clinical trial, doctors have begun transplanting fetal pig neurons that produce GABA into the brains of patients to learn whether the cell transplants can help control seizures. Preliminary research suggests that stem cell transplants also may prove beneficial for treating epilepsy. Research showing that the brain undergoes subtle changes prior to a seizure has led to a prototype device that may be able to predict seizures up to 3 minutes before they begin. If this device works, it could greatly reduce the risk of injury from seizures by allowing people to move to a safe area before their seizures start. This type of device also may be hooked up to a treatment pump or other device that will automatically deliver an antiepileptic drug or an electric impulse to forestall the seizures. Researchers are continually improving MRI and other brain scans. Pre-surgical brain imaging can guide doctors to abnormal brain tissue and away from essential parts of the brain. Researchers also are using brain scans such as magnetoencephalograms (MEG) and magnetic resonance spectroscopy (MRS) to identify and study subtle problems in the brain that cannot otherwise be detected. Their findings may lead to a better understanding of epilepsy and how it can be treated. How Can I Help Research on Epilepsy?There are many ways that people with epilepsy and their families can help with research on this disorder. Pregnant women with epilepsy who are taking antiepileptic drugs can help researchers learn how these drugs affect unborn children by participating in the Antiepileptic Drug Pregnancy Registry, which is maintained by the Genetics and Teratology Unit of Massachusetts General Hospital (see section on Pregnancy and Motherhood). People with epilepsy that may be hereditary can aid research by participating in the Epilepsy Gene Discovery Project, which is supported by the Epilepsy Foundation. This project helps to educate people with epilepsy about new genetic research on the disorder and enlists families with hereditary epilepsy for participation in gene research. People who enroll in this project are asked to create a family tree showing which people in their family have or have had epilepsy. Researchers then examine this information to determine if the epilepsy is in fact hereditary, and they may invite participants to enroll in genetic research studies. In many cases, identifying the gene defect responsible for epilepsy in an individual family leads researchers to new clues about how epilepsy develops. It also can provide opportunities for early diagnosis and genetic screening of individuals in the family. People with epilepsy can help researchers test new medications, surgical techniques, and other treatments by enrolling in clinical trials. Information on clinical trials can be obtained from the NINDS as well as many private pharmaceutical and biotech companies, universities, and other organizations. A person who wishes to participate in a clinical trial must ask his or her regular physician to refer him or her to the doctor in charge of that trial and to forward all necessary medical records. While experimental therapies may benefit those who participate in clinical trials, patients and their families should remember that all clinical trials also involve some risks. Therapies being tested in clinical trials may not work, and in some cases doctors may not yet be sure that the therapies are safe. Patients should be certain they understand the risks before agreeing to participate in a clinical trial. Patients and their families also can help epilepsy research by donating their brain to a brain bank after death. Brain banks supply researchers with tissue they can use to study epilepsy and other disorders. Below are some brain banks that accept tissue from patients with epilepsy: Brain and Tissue Bank for Developmental Disorders (tissue from children only) (tissue from adults only) National Disease Research Interchange Human Brain and Spinal Fluid Resource Center What To Do If You See Someone Having a SeizureIf you see someone having a seizure with convulsions and/or loss of consciousness, here's how you can help:
Call 911 if: The person is pregnant or has diabetes. The seizure happened in water. The seizure lasts longer than 5 minutes. The person does not begin breathing again or does not return to consciousness after the seizure stops. Another seizure starts before the person regains consciousness. The person injures himself or herself during the seizure. This is a first seizure or you think it might be. If in doubt, check to see if the person has a medical identification card or jewelry stating that they have epilepsy or a seizure disorder. After the seizure ends, the person will probably be groggy and tired. He or she also may have a headache and be confused or embarrassed. Be patient with the person and try to help him or her find a place to rest if he or she is tired or doesn't feel well. If necessary, offer to call a taxi, a friend, or a relative to help the person get home safely. If you see someone having a non-convulsive seizure, remember that the person's behavior is not intentional. The person may wander aimlessly or make alarming or unusual gestures. You can help by following these guidelines: Remove any dangerous objects from the area around the person or in his or her path. Don't try to stop the person from wandering unless he or she is in danger. Don't shake the person or shout. Stay with the person until he or she is completely alert. ConclusionMany people with epilepsy lead productive and outwardly normal lives. Medical and research advances in the past two decades have led to a better understanding of epilepsy and seizures than ever before. Advanced brain scans and other techniques allow greater accuracy in diagnosing epilepsy and determining when a patient may be helped by surgery. More than 20 different medications and a variety of surgical techniques are now available and provide good control of seizures for most people with epilepsy. Other treatment options include the ketogenic diet and the first implantable device, the vagus nerve stimulator. Research on the underlying causes of epilepsy, including identification of genes for some forms of epilepsy and febrile seizures, has led to a greatly improved understanding of epilepsy that may lead to more effective treatments or even new ways of preventing epilepsy in the future. Information Resources: March 2004Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
Note: Due to the large number of epilepsy syndromes and treatments, only a few are discussed in this booklet. Additional information may be available from your doctor, other health professionals, medical libraries, or by calling the NINDS Office of Communications and Public Liaison at the number provided on the Information Resources card in the back pocket of this brochure.
NIH Publication No. 04-156 |
|
Introduction
What is Shingles? Who is at Risk for Shingles? What are the Symptoms of Shingles? How Should Shingles Be Treated? Is Shingles Contagious? Can Shingles Be Prevented? What is Postherpetic Neuralgia? What are Other Complications of Shingles? Can Infection with VZV During Pregnancy Harm the Baby? What Research is Being Done? “On Catching Chickenpox . . . but not Catching Shingles” Where can I get more information? Glossary IntroductionWhen the itchy red spots of childhood chickenpox* disappear and life returns to normal, the battle with the virus that causes chickenpox seems won. But for too many of us this triumph of immune system over virus is temporary. The virus has not been destroyed but remains dormant in our nerve cells, ready to strike again later in life. This second eruption of the chickenpox virus is the disease called shingles or herpes-zoster . "I was having exams at college and I got a rash in a band around one side of my waist. The spots were very painful. At first I thought it was chickenpox, but I'd had that years before," recalls a young woman who had shingles in her twenties. The young woman's memory was correct. She did have chickenpox as a child. You cannot develop shingles unless you have had an earlier exposure to chickenpox, and most people who get chickenpox are at risk for shingles. The woman had the typical one-sided band of rash and pain of this common neurological disorder. Her age was unusual, however. While young people do develop shingles, the disease most often strikes after age 40. But since shingles is so common, affecting an estimated one-quarter of Americans at some point during their lifetimes, cases in young people are not rare. * Terms in Italics are defined in the Glossary. What is Shingles?Scientists
call the virus that causes chickenpox/shingles varicella-zoster
virus or VZV. The word "varicella" is derived from "variola," the
Latin word for smallpox, another infectious disease that can resemble
chickenpox. (Smallpox is a highly contagious and often fatal disease
that has disfigured or killed millions of people, especially during
the Middle Ages.) “Zoster” is the Greek word for girdle;
shingles often produces a girdle or belt of blisters or lesions
around one side of the waist. This striking pattern also underlies
the condition's common name: shingles comes from “cingulum,” the
Latin word for belt or girdle.
VZV belongs to a group of viruses called herpesviruses. This group includes the herpes simplex virus that causes cold sores, fever blisters, mononucleosis, genital herpes (a sexually transmitted disease), and Epstein-Barr virus involved in infectious mononucleosis. Like VZV, other herpesviruses can hide in the nervous system after an initial infection and then travel down nerve cell fibers to cause a renewed infection. Repeated episodes of cold sores on the lips are the most common example. As early as 1909, scientists suspected that the viruses causing chickenpox and shingles were one and the same. In the 1920s and 1930s, the case was strengthened by an experiment in which children were inoculated with fluid from shingles blisters. Within 2 weeks, about half of the children developed chickenpox. Finally, in 1958, detailed analyses of the viruses taken from patients with either chickenpox or shingles confirmed that the viruses were identical. Virtually all adults in the United States have had chickenpox, even if it was so mild as to pass unnoticed, and thus may develop shingles later in life. In the original exposure to VZV (chickenpox), some of the virus particles leave the blood and settle into clusters of nerve cells (neurons ) called sensory ganglia, where they remain for many years in an inactive (latent) form. The sensory ganglia, which are adjacent to the spinal cord and brain, relay information to the brain about what the body is sensing - heat, cold, touch, pain. When the VZV reactivates, it spreads down the long nerve fibers (axons) that extend from the sensory cell bodies to the skin. The viruses multiply, the telltale rash erupts, and the person now has herpes-zoster, or shingles. With shingles, the nervous system is more deeply involved than it was during the bout with chickenpox, and the symptoms are often more complex and severe. Who is at Risk for Shingles?About 25 percent of all adults, mostly otherwise healthy, will get shingles during their lifetimes, usually after age 40. The incidence increases with age so that shingles is 10 times more likely to occur in adults over 60 than in children under 10. People with compromised immune systems - from use of immunosuppressive medications such as prednisone, from serious illnesses such as cancer, or from infection with HIV - are at special risk of developing shingles. These individuals also can have re-eruptions and some may have shingles that never heals. Most people who get shingles re-boost their immunity to VZV and will not get the disease for another few decades. Youngsters whose mothers had chickenpox late in pregnancy - 5 to 21 days before giving birth - or who had chickenpox in infancy, have an increased risk of pediatric shingles. Sometimes these children are born with chickenpox or develop a typical case within a few days (see section entitled "Can Shingles During Pregnancy Harm the Baby?" for more information). What are the Symptoms of Shingles?The first sign of shingles is often burning or tingling pain, or itch, in one particular location on only one side of the body. After several days or a week, a rash of fluid-filled blisters, similar to chickenpox, appears in one area on one side of the body. Recent studies have shown that subtle cases of shingles with only a few lesions, or none, are more common than previously thought. These cases will usually remain unrecognized. Cases without any known lesions are known as zoster sine herpete. Shingles pain can be mild or intense. Some people have mostly itching; some feel pain from the gentlest touch or breeze. The most common location for shingles is a band, called a dermatome, spanning one side of the trunk around the waistline. The second most common location is on one side of the face around the eye and on the forehead. However, shingles can involve any part of the body. The number of lesions is variable. Some rashes merge and produce an area that looks like a severe burn. Other patients may have just a few scattered lesions that don't cause severe symptoms. For most healthy people, shingles rashes heal within a few weeks, the pain and itch that accompany the lesions subside, and the blisters leave no scars. Other people may have sensory symptoms that linger for a few months. How Should Shingles Be Treated?Shingles attacks can be made less severe and shorter by using prescription antiviral drugs: acyclovir, valacyclovir, or famcyclovir. Acyclovir is available in a generic form, but the pills must be taken five times a day, whereas valacyclovir and famcyclovir pills are taken three times a day. It is important not to miss any doses and not to stop taking the medication early. Antiviral drugs can reduce by about half the risk of being left with postherpetic neuralgia (see section entitled "What is Postherpetic Neuralgia?"), which is chronic pain that can last for months or years after the shingles rash clears. Doctors recommend starting antiviral drugs at the first sign of the shingles rash, or even if the telltale symptoms indicate that a rash is about to erupt. Even if a patient is not seen by a doctor at the beginning of the illness, it may still be useful to start antiviral medications if new lesions are still forming. Other treatments to consider are anti-inflammatory corticosteroids such as prednisone. These are routinely used when the eye or other facial nerves are affected. Is Shingles Contagious?A person with a shingles rash can pass the virus to someone, usually a child, who has never had chickenpox, but the child will develop chickenpox, not shingles. The child must come into direct contact with the open sores of the shingles rash. Merely being in the same room with a shingles patient will not cause the child to catch chickenpox because during a shingles infection the virus is not normally in the lungs and therefore can't be spread through the air. People with chickenpox cannot communicate shingles to someone else although they can of course pass the chickenpox on to someone who has never had chickenpox. In cases of chickenpox, the virus can become airborne because it is found in the upper respiratory tract. Shingles occurs when an unknown trigger causes the virus hiding inside the person's body to become activated. Unlike chickenpox, shingles can't be "caught" from someone else. Can Shingles Be Prevented?Chickenpox vaccine Immunization with the varicella vaccine (or chickenpox vaccine) - now recommended in the United States for all children between 18 months and adolescence - can protect children from getting chickenpox. People who have been vaccinated against chickenpox are less likely to get shingles because the weak, “attenuated” strain of virus used in the chickenpox vaccine is less likely to survive in the body over decades. But a definitive answer to the question of whether shingles can occur later in life in a person vaccinated against chickenpox will only be provided when enough data have been gathered over the next several decades. Some scientists believe that immunizing children against chickenpox increases the risk of shingles in adults who were not themselves immunized during childhood. This is because when adults care for children sick with chickenpox, it reboosts their own immunity that keeps the virus in their nerve cells from reactivating as shingles. With fewer children coming down with chickenpox, there are fewer opportunities for this "reboosting" of adult immunity, and so there may be more shingles cases for the next 40-50 years. Shingles vaccine In May 2006, the Food and Drug Administration approved a VZV vaccine (Zostavax) for use in people 60 and older who have had chickenpox. When the vaccine becomes more widely available, many older adults will for the first time have a means of preventing shingles. Researchers found that giving older adults the vaccine reduced the expected number of cases of shingles by half. And in people who still got the disease despite immunization, the severity and complications of shingles were dramatically reduced. The Shingles Prevention Study - a collaboration between the Department of Veterans Affairs, the National Institute of Allergy and Infectious Diseases, and Merck & Co., Inc. - involved more than 38,000 veterans aged 60 and older. The purpose was to find out how safe the vaccine is, and if it can prevent shingles. Half the study participants were vaccinated with a more potent version of the chickenpox vaccine, developed specifically for use in adults, and half received a placebo vaccine. Neither volunteers nor researchers knew if a particular subject had gotten active or placebo vaccine until after the end of the study (a double-blind study). During more than 3 years of follow up, the vaccine reduced shingles cases by 51 percent; 642 cases of shingles developed in the placebo group compared with only 315 in the vaccinated group. Pain and discomfort were reduced by 61 percent in people who received the active vaccine but still got shingles. The vaccine also reduced the number of cases of postherpetic neuralgia by two-thirds compared with the placebo. The shingles vaccine is only a preventive therapy and not a treatment for those who already have shingles or postherpetic neuralgia. What is Postherpetic Neuralgia?Sometimes, particularly in older people, shingles pain persists long after the rash has healed. This postherpetic neuralgia can be mild or severe - the most severe cases can lead to insomnia, weight loss, depression, and disability. Postherpetic neuralgia is not directly life-threatening. About a dozen medications in four categories have been shown in clinical trials to provide some pain relief. These include: Tricyclic antidepressants (TCAs): TCAs are often the first type of drug given to patients suffering from postherpetic neuralgia. The TCA amitryptiline was commonly prescribed in the past, but although effective, it has a high rate of side effects. Desipramine and nortriptyline have fewer side effects and are therefore better choices for older adults, the most likely group to have postherpetic neuralgia. Common side effects of TCAs include dry eyes and mouth, constipation, and grogginess. People with heart arrhythmias, previous heart attacks, or narrow angle glaucoma should usually use a different class of drugs. Anticonvulsants: Some drugs that reduce seizures can also treat postherpetic neuralgia because seizures and pain both involve abnormally increased firing of nerve cells. An antiseizure medication, carbamazepine, is effective for postherpetic neuralgia but has rare, potentially dangerous side effects so a newer anticonvulsant, gabapentin, is far more often prescribed. Side effects of the drug include drowsiness or confusion, dizziness, and sometimes ankle swelling. Opioids: Opioids are strong pain medications used for all types of pain. They include oxycodone, morphine, tramadol, and methadone. Opioids can have side effects - including drowsiness, mental dulling, and constipation - and can be addictive, so their use must be monitored carefully in those with a history of addiction. Topical local anesthetics: Local anesthetics applied directly to the skin of the painful area affected by postherpetic neuralgia are also effective. Lidocaine, the most commonly prescribed, is available in cream, gel, or spray form. It is also available in a patch that has been approved by the Food and Drug Administration for use specifically in postherpetic neuralgia. With topical local anesthetics, the drug stays in the skin and therefore does not cause problems such as drowsiness or constipation. Capsaicin cream may be somewhat effective and is available over the counter, but most people find that it causes severe burning pain during application. Postherpetic itchThe itch that sometimes occurs during or after shingles can be quite severe and painful. Clinical experience suggests that postherpetic itch is harder to treat than postherpetic neuralgia. Topical local anesthetics (which numb the skin) provide substantial relief to some patients. Since postherpetic itch typically develops in skin that has severe sensory loss, it is particularly important to avoid scratching. Scratching numb skin too long or too hard can cause injury. What are Other Complications of Shingles?People with ophthalmic shingles -- lesions in or around the eye and forehead -- can suffer painful eye infections, and in some cases immediate or delayed vision loss. People with shingles in or near the eye should see an ophthalmologist immediately. Shingles infections within or near the ear (Ramsay-Hunt syndrome) can cause hearing or balance problems as well as weakness of the muscles on the affected side of the face. In rare cases, shingles can spread into the brain or spinal cord and cause serious complications such as stroke or meningitis (an infection of the membranes outside the brain and spinal cord). People with shingles need to seek immediate medical evaluation if they notice neurological symptoms outside the region of the primary shingles attack. People who are immunosuppressed, whether from diseases such as HIV or medications, have an increased risk of serious complications from shingles. Most commonly, they get shingles that spreads to involve more parts of the body, or shingles rashes that persist for long periods or return frequently. Many such patients are helped by taking antiviral medications on a continuous basis. Can Infection with VZV During Pregnancy Harm the Baby?Many mothers-to-be are concerned about any infection contracted during pregnancy, and rightly so because some infections can be transmitted across the mother's bloodstream to the fetus or can be acquired by the baby during the birth process. VZV infection during pregnancy poses some risk to the unborn child, depending upon the stage of pregnancy. During the first 30 weeks, maternal chickenpox may, in some cases, lead to congenital malformations. Such cases are rare and experts differ in their opinions on how great the risk is. Most experts agree that shingles in a pregnant woman, a rare event, is even less likely to cause harm to the unborn child. If a pregnant woman gets chickenpox between 21 to 5 days before giving birth, her newborn can have chickenpox at birth or develop it within a few days. But the time lapse between the start of the mother's illness and the birth of the baby generally allows the mother's immune system to react and produce antibodies to fight the virus. These antibodies can be transmitted to the unborn child and thus help fight the infection. Still, a small percent of the babies exposed to chickenpox in the 21 to 5 days before birth develop shingles in the first 5 years of life because the newborn's immune system is not yet fully functional and capable of keeping the virus latent. What if the mother contracts chickenpox at the time of birth? In that case the mother's immune system has not had a chance to mobilize its forces. And although some of the mother's antibodies will be transmitted to the newborn via the placenta, the newborn will have little ability to fight off the attack because its immune system is immature. If these babies develop chickenpox as a result, it can be fatal. They are given zoster immune globulin, a preparation made from the antibody-rich blood of adults who have recently recovered from chickenpox or shingles, to lessen the severity of their chickenpox. What Research is Being Done?Because of nervous system involvement, the chickenpox/shingles virus is studied by the National Institute of Neurological Disorders and Stroke, a part of the National Institutes of Health. The National Institute of Allergy and Infectious Diseases, the National Cancer Institute, the National Institute on Aging, and the National Eye Institute also support research on shingles. Medical research on shingles has two main goals. The first is to develop drugs to fight the disease and to prevent or treat its complications, especially postherpetic neuralgia. The second is to understand the disease well enough to prevent it, especially in people at high risk. Scientists need to learn much more about the VZV, particularly how it becomes latent in the body and what induces it to become active again. Scientists suspect that the VZV DNA is inserted into one of the chromosomes of the nerve cell - the units that house the cell's own genetic material. A healthy immune system protects against all kinds of diseases, but people with faulty immunity are vulnerable to many illnesses, including shingles. Antibodies, one of the immune system's major defense mechanisms against infection, are not very helpful against shingles. The immune cells that appear to combat shingles are two types of white blood cells: T lymphocytes and macrophages. Scientists are trying to find ways to boost the activity of these cells - especially in patients at high risk for severe or disseminated shingles (a rare condition in which the virus spreads to other areas of the body, sometimes vital areas such as the blood or the lungs). Other researchers are studying how VZV infects neurons. In particular, they are looking at how the virus assembles in and exits out of nerve cells, with the goal of blocking this important step. In another study, researchers are developing animal models to evaluate VZV vaccines. Their findings may lead to improved vaccines that protect against varicella or prevent it from establishing latent infection or reactivation to cause shingles and postherpetic neuralgia. Other research is aimed at finding new methods for identifying the biological differences between people who suffer from or escape long-term postherpetic neuralgia pain after shingles. The goals of this research are to identify ways to reduce the risk of postherpetic neuralgia after shingles. “On Catching Chickenpox . . . but not Catching Shingles”Chickenpox and shingles are caused by the same virus - varicella-zoster (VZV). When a person, usually a child, who has not received the chickenpox vaccine (which became available in the United States in 1995) is exposed to VZV, he or she usually develops chickenpox, a highly contagious disease that can be spread by breathing as well as by contact with the rash. The infection begins in the upper respiratory tract where the virus incubates for 15 days or more. VZV then spreads to the bloodstream and migrates to the skin, giving rise to the familiar chickenpox rash. In contrast, you can't catch shingles from someone else. You must already have been exposed to chickenpox and harbor the virus in your nervous system to develop shingles. When reactivated, the virus travels down nerves to the skin, causing the painful shingles rash. In shingles, the virus does not normally spread to the bloodstream or lungs, so the virus is not shed in air. Because the shingles rash contains active virus particles, someone who has never had chickenpox can catch it from exposure to a shingles rash. Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
acyclovir - one of three available antiviral drugs that can reduce the severity and duration of a shingles attack if given soon after onset. capsaicin - an active ingredient in hot chili peppers used in topical ointments to relieve pain. It appears to work by reducing a chemical substance found at nerve endings and involved in transmitting pain signals to the brain. While somewhat effective for postherpetic neuralgia, it can cause severe burning in some patients. carbamazepine - a drug that works both as an anticonvulsant and a pain reliever. chickenpox - an acute contagious disease that usually occurs in children and is caused by the varicella-zoster virus. desipramine - an antidepressant often prescribed to help reduce the pain from postherpetic neuralgia. Doctors often prescribe it because it has fewer side effects than some other antidepressants. famcyclovir - one of three available antiviral drugs that can reduce the severity and duration of a shingles attack if given soon after onset. gabapentin -- an antiseizure medicine that is also used as a pain reliever. herpes zoster - the medical term for shingles; an infection caused by the varicella-zoster virus, one of the herpesviruses family of viruses. herpes simplex - the medical term for a related but different virus that causes repeated mild blisters of the skin or mucous membrane. Herpes simplex rashes can return many times, whereas shingles usually appears no more than once or twice in a person's lifetime. herpesviruses - a large family of viruses that cause a number of related conditions including, but not limited to, oral and genital herpes simplex, varicella (chickenpox), and herpes-zoster (shingles). immunosuppressed - having a weakened immune system. Common causes are certain illnesses (HIV, some cancers) or use of certain drugs such as prednisone. latent - hidden, dormant, inactive. The virus that causes chickenpox remains hidden in the nervous system after the initial attack of chickenpox is over. When it becomes reactivated, usually many years later, the virus can cause shingles. lidocaine - a pain-killing drug sometimes used for treating postherpetic neuralgia. It is available in an adhesive fabric patch that can be placed on the skin directly over the site of the pain. neuron- the functional cell of the brain and nervous system. nortriptyline- an antidepressant often prescribed to help reduce the pain from postherpetic neuralgia. Doctors often prescribe it because it has fewer side effects than some other antidepressants. postherpetic itch - severe, painful, and difficult to treat itching that sometimes accompanies postherpetic neuralgia. Topical local anesthetics provide relief to some patients. postherpetic neuralgia - a condition characterized by pain that persists more than 3 months after healing of a shingles rash; caused by damage to the nervous system. prednisone - an anti-inflammatory corticosteroid drug routinely given to shingles patients when an eye or other facial nerve is involved. valacyclovir - one of three available antiviral drugs that can reduce the severity and duration of a shingles attack if given soon after onset. varicella-zoster virus - a virus that causes two distinct diseases, chickenpox and shingles. It is a member of the herpesvirus family. "Varicella" is Latin for little pox; "zoster" is the Greek word for girdle. Medically, zoster is sometimes used as a synonym for shingles. zoster sine herpete - a case of shingles in which there are no blisters or other signs of the illness on the skin. NIH Publication No. 06-307
|
|
A
Short History of the Treatment of Spinal Cord Injury
What Is a Spinal Cord Injury? How Does the Spinal Cord Work? What Happens When the Spinal Cord Is Injured? What Are the Immediate Treatments for Spinal Cord Injury? How Does a Spinal Cord Injury Affect the Rest of the Body? How Does Rehabilitation Help People Recover From Spinal Cord Injuries? How Is Research Helping Spinal Cord Injury Patients? The Future of Spinal Cord Research Where can I get more information? Glossary A Short History of the Treatment of Spinal Cord InjuryAccounts of spinal cord injuries and their treatment date back to ancient times, even though there was little chance of recovery from such a devastating injury. The earliest is found in an Egyptian papyrus roll manuscript written in approximately 1700 B.C. that describes two spinal cord injuries involving fracture or dislocation of the neck vertebrae accompanied by paralysis.* The description of each was "an ailment not to be treated." Centuries later in Greece, treatment for spinal cord injuries had changed little. According to the Greek physician Hippocrates (460-377 B.C.) there were no treatment options for spinal cord injuries that resulted in paralysis; unfortunately, those patients were destined to die. But Hippocrates did use rudimentary forms of traction to treat spinal fractures without paralysis. The Hippocratic Ladder was a device that required the patient to be bound, tied to the rungs upside-down, and shaken vigorously to reduce spinal curvature. Another invention, the Hippocratic Board, allowed the doctor to apply traction to the immobilized patient's back using either his hands and feet or a wheel and axle arrangement. Hindu, Arab, and Chinese physicians also developed basic forms of traction to correct spinal deformities. These same principles of traction are still applied today. In about 200 A.D., the Roman physician Galen introduced the concept of the central nervous system when he proposed that the spinal cord was an extension of the brain that carried sensation to the limbs and back. By the seventh century A.D., Paulus of Aegina was recommending surgery for spinal column fracture to remove the bone fragments that he was convinced caused paralysis. In his influential anatomy textbook published in 1543, the Renaissance physician and teacher Vesalius described and illustrated the spinal cord in all its parts. The illustrations in his books, based on direct observation and dissection of the spine, gave physicians a way to understand the basic structure of the spine and spinal cord and what could happen when it was injured. The words we use today to identify segments of the spine - cervical, thoracic, lumbar, sacral, and coccygeal - come directly from Vesalius. With the widespread use of antiseptics and sterilization in surgical procedures in the late nineteenth century, spinal surgery could finally be done with a much lower risk of infection. The use of X-rays, beginning in the 1920s, gave surgeons a way to precisely locate the injury and also made diagnosis and prediction of outcome more accurate. By the middle of the twentieth century, a standard method of treating spinal cord injuries was established - reposition the spine, fix it in place, and rehabilitate disabilities with exercise. In the 1990s, the discovery that the steroid drug methylprednisolone could reduce damage to nerve cells if given early enough after injury gave doctors an additional treatment option. What Is a Spinal Cord Injury?Although the hard bones of the spinal column protect the soft tissues of the spinal cord, vertebrae can still be broken or dislocated in a variety of ways and cause traumatic injury to the spinal cord. Injuries can occur at any level of the spinal cord. The segment of the cord that is injured, and the severity of the injury, will determine which body functions are compromised or lost. Because the spinal cord acts as the main information pathway between the brain and the rest of the body, a spinal cord injury can have significant physiological consequences. Catastrophic falls, being thrown from a horse or through a windshield, or any kind of physical trauma that crushes and compresses the vertebrae in the neck can cause irreversible damage at the cervical level of the spinal cord and below. Paralysis of most of the body including the arms and legs, called quadriplegia, is the likely result. Automobile accidents are often responsible for spinal cord damage in the middle back (the thoracic or lumbar area), which can cause paralysis of the lower trunk and lower extremities, called paraplegia. Other kinds of injuries that directly penetrate the spinal cord, such as gunshot or knife wounds, can either completely or partially sever the spinal cord and create life-long disabilities. Most injuries to the spinal cord don't completely sever it. Instead, an injury is more likely to cause fractures and compression of the vertebrae, which then crush and destroy the axons, extensions of nerve cells that carry signals up and down the spinal cord between the brain and the rest of the body. An injury to the spinal cord can damage a few, many, or almost all of these axons. Some injuries will allow almost complete recovery. Others will result in complete paralysis. Until World War II, a serious spinal cord injury usually meant certain death, or at best a lifetime confined to a wheelchair and an ongoing struggle to survive secondary complications such as breathing problems or blood clots. But today, improved emergency care for people with spinal cord injuries and aggressive treatment and rehabilitation can minimize damage to the nervous system and even restore limited abilities. Advances in research are giving doctors and patients hope that all spinal cord injuries will eventually be repairable. With new surgical techniques and exciting developments in spinal nerve regeneration, the future for spinal cord injury survivors looks brighter every day. This brochure has been written to explain what happens to the spinal cord when it is injured, the current treatments for spinal cord injury patients, and the most promising avenues of research currently under investigation. Facts and Figures About Spinal Cord Injury
How Does the Spinal Cord Work?
To understand what can happen as the result of a spinal cord injury, it helps to know the anatomy of the spinal cord and its normal functions. Spine Anatomy The soft, jelly-like spinal cord is protected by the spinal column. The spinal column is made up of 33 bones called vertebrae, each with a circular opening similar to the hole in a donut. The bones are stacked one on top of the other and the spinal cord runs through the hollow channel created by the holes in the stacked bones. The vertebrae can be organized into sections, and are named and numbered from top to bottom according to their location along the backbone:
Although the hard vertebrae protect the soft spinal cord from injury most of the time, the spinal column is not all hard bone. Between the vertebrae are discs of semi-rigid cartilage, and in the narrow spaces between them are passages through which the spinal nerves exit to the rest of the body. These are places where the spinal cord is vulnerable to direct injury. The spinal cord is also organized into segments and named and numbered from top to bottom. Each segment marks where spinal nerves emerge from the cord to connect to specific regions of the body. Locations of spinal cord segments do not correspond exactly to vertebral locations, but they are roughly equivalent.
The single coccygeal nerve carries sensory information from the skin of the lower back. Spinal Cord Anatomy The spinal cord has a core of tissue containing nerve cells, surrounded by long tracts of nerve fibers consisting of axons. The tracts extend up and down the spinal cord, carrying signals to and from the brain. The average size of the spinal cord varies in circumference along its length from the width of a thumb to the width of one of the smaller fingers. The spinal cord extends down through the upper two thirds of the vertebral canal, from the base of the brain to the lower back, and is generally 15 to 17 inches long depending on an individual's height. The interior of the spinal cord is made up of neurons, their support cells called glia, and blood vessels. The neurons and their dendrites (branching projections that help neurons communicate with each other) reside in an H-shaped region called "grey matter." The H-shaped grey matter of the spinal cord contains motor neurons that control movement, smaller interneurons that handle communication within and between the segments of the spinal cord, and cells that receive sensory signals and then send information up to centers in the brain. Surrounding the grey matter of neurons is white matter. Most axons are covered with an insulating substance called myelin, which allows electrical signals to flow freely and quickly. Myelin has a whitish appearance, which is why this outer section of the spinal cord is called "white matter." Axons carry signals downward from the brain (along descending pathways) and upward toward the brain (along ascending pathways) within specific tracts. Axons branch at their ends and can make connections with many other nerve cells simultaneously. Some axons extend along the entire length of the spinal cord. The descending motor tracts control the smooth muscles of internal organs and the striated (capable of voluntary contractions) muscles of the arms and legs. They also help adjust the autonomic nervous system's regulation of blood pressure, body temperature, and the response to stress. These pathways begin with neurons in the brain that send electrical signals downward to specific levels of the spinal cord. Neurons in these segments then send the impulses out to the rest of the body or coordinate neural activity within the cord itself. The ascending sensory tracts transmit sensory signals from the skin, extremities, and internal organs that enter at specific segments of the spinal cord. Most of these signals are then relayed to the brain. The spinal cord also contains neuronal circuits that control reflexes and repetitive movements, such as walking, which can be activated by incoming sensory signals without input from the brain. The circumference of the spinal cord varies depending on its location. It is larger in the cervical and lumbar areas because these areas supply the nerves to the arms and upper body and the legs and lower body, which require the most intense muscular control and receive the most sensory signals. The ratio of white matter to grey matter also varies at each level of the spinal cord. In the cervical segment, which is located in the neck, there is a large amount of white matter because at this level there are many axons going to and from the brain and the rest of the spinal cord below. In lower segments, such as the sacral, there is less white matter because most ascending axons have not yet entered the cord, and most descending axons have contacted their targets along the way. To pass between the vertebrae, the axons that link the spinal cord to the muscles and the rest of the body are bundled into 31 pairs of spinal nerves, each pair with a sensory root and a motor root that make connections within the grey matter. Two pairs of nerves - a sensory and motor pair on either side of the cord - emerge from each segment of the spinal cord. The functions of these nerves are determined by their location in the spinal cord. They control everything from body functions such as breathing, sweating, digestion, and elimination, to gross and fine motor skills, as well as sensations in the arms and legs. The Nervous Systems Together, the spinal cord and the brain make up the central nervous system (CNS). The CNS controls most functions of the body, but it is not the only nervous system in the body. The peripheral nervous system (PNS) includes the nerves that project to the limbs, heart, skin, and other organs outside the brain. The PNS controls the somatic nervous system, which regulates muscle movements and the response to sensations of touch and pain, and the autonomic nervous system, which provides nerve input to the internal organs and generates automatic reflex responses. The autonomic nervous system is divided into the sympathetic nervous system, which mobilizes organs and their functions during times of stress and arousal, and the parasympathetic nervous system, which conserves energy and resources during times of rest and relaxation. The spinal cord acts as the primary information pathway between the brain and all the other nervous systems of the body. It receives sensory information from the skin, joints, and muscles of the trunk, arms, and legs, which it then relays upward to the brain. It carries messages downward from the brain to the PNS, and contains motor neurons, which direct voluntary movements and adjust reflex movements. Because of the central role it plays in coordinating muscle movements and interpreting sensory input, any kind of injury to the spinal cord can cause significant problems throughout the body. What Happens When the Spinal Cord Is Injured?A spinal cord injury usually begins with a sudden, traumatic blow to the spine that fractures or dislocates vertebrae. The damage begins at the moment of injury when displaced bone fragments, disc material, or ligaments bruise or tear into spinal cord tissue. Axons are cut off or damaged beyond repair, and neural cell membranes are broken. Blood vessels may rupture and cause heavy bleeding in the central grey matter, which can spread to other areas of the spinal cord over the next few hours. Within minutes, the spinal cord swells to fill the entire cavity of the spinal canal at the injury level. This swelling cuts off blood flow, which also cuts off oxygen to spinal cord tissue. Blood pressure drops, sometimes dramatically, as the body loses its ability to self-regulate. As blood pressure lowers even further, it interferes with the electrical activity of neurons and axons. All these changes can cause a condition known as spinal shock that can last from several hours to several days. Although there is some controversy among neurologists about the extent and impact of spinal shock, and even its definition in terms of physiological characteristics, it appears to occur in approximately half the cases of spinal cord injury, and it is usually directly related to the size and severity of the injury. During spinal shock, even undamaged portions of the spinal cord become temporarily disabled and can't communicate normally with the brain. Complete paralysis may develop, with loss of reflexes and sensation in the limbs. The crushing and tearing of axons is just the beginning of the devastation that occurs in the injured spinal cord and continues for days. The initial physical trauma sets off a cascade of biochemical and cellular events that kills neurons, strips axons of their myelin insulation, and triggers an inflammatory immune system response. Days or sometimes even weeks later, after this second wave of damage has passed, the area of destruction has increased - sometimes to several segments above and below the original injury - and so has the extent of disability.
What Are the Immediate Treatments for Spinal Cord Injury?The outcome of any injury to the spinal cord depends upon the number of axons that survive: the higher the number of normally functioning axons, the less the amount of disability. Consequently, the most important consideration when moving people to a hospital or trauma center is preventing further injury to the spine and spinal cord. Spinal cord injury isn't always obvious. Any injury that involves the head (especially with trauma to the front of the face), pelvic fractures, penetrating injuries in the area of the spine, or injuries that result from falling from heights should be suspect for spinal cord damage. Until imaging of the spine is done at an emergency or trauma center, people who might have spinal cord injury should be cared for as if any significant movement of the spine could cause further damage. They are usually transported in a recumbent (lying down) position, with a rigid collar and backboard immobilizing the spine. Respiratory complications are often an indication of the severity of spinal cord injury. About one third of those with injury to the neck area will need help with breathing and require respiratory support via intubation, which involves inserting a tube connected to an oxygen tank through the nose or throat and into the airway. Methylprednisolone, a steroid drug, became standard treatment for acute spinal cord injury in 1990 when a large-scale clinical trial supported by the National Institute of Neurological Disorders and Stroke showed significantly better recovery in patients who were given the drug within the first 8 hours after their injury. Methylprednisolone appears to reduce the damage to nerve cells and decreases inflammation near the injury site by suppressing activities of immune cells. Realignment of the spine using a rigid brace or axial traction is usually done as soon as possible to stabilize the spine and prevent additional damage. On about the third day after the injury, doctors give patients a complete neurological examination to diagnose the severity of the injury and predict the likely extent of recovery. The ASIA Impairment Scale is the standard diagnostic tool used by doctors. X-rays, MRIs, or more advanced imaging techniques are also used to visualize the entire length of the spine. ASIA (American Spinal Injury Association) Impairment Scale*
* Used with permission of the American Spinal Injury Association. Spinal cord injuries are classified as either complete or incomplete, depending on how much cord width is injured. An incomplete injury means that the ability of the spinal cord to convey messages to or from the brain is not completely lost. People with incomplete injuries retain some motor or sensory function below the injury. A complete injury is indicated by a total lack of sensory and motor function below the level of injury. How Does a Spinal Cord Injury Affect the Rest of the Body?People who survive a spinal cord injury will most likely have medical complications such as chronic pain and bladder and bowel dysfunction, along with an increased susceptibility to respiratory and heart problems. Successful recovery depends upon how well these chronic conditions are handled day to day.
How Does Rehabilitation Help People Recover From Spinal Cord Injuries?No two people will experience the same emotions after surviving a spinal cord injury, but almost everyone will feel frightened, anxious, or confused about what has happened. It's common for people to have very mixed feelings: relief that they are still alive, but disbelief at the nature of their disabilities. Rehabilitation programs combine physical therapies with skill-building activities and counseling to provide social and emotional support. The education and active involvement of the newly injured person and his or her family and friends is crucial. A rehabilitation team is usually led by a doctor specializing in physical medicine and rehabilitation (called a physiatrist), and often includes social workers, physical and occupational therapists, recreational therapists, rehabilitation nurses, rehabilitation psychologists, vocational counselors, nutritionists, and other specialists. A case-worker or program manager coordinates care. In the initial phase of rehabilitation, therapists emphasize regaining leg and arm strength since mobility and communication are the two most important areas of function. For some, mobility will only be possible with the assistance of devices such as a walker, leg braces, or a wheelchair. Communication skills, such as writing, typing, and using the telephone, may also require adaptive devices. Physical therapy includes exercise programs geared toward muscle strengthening. Occupational therapy helps redevelop fine motor skills. Bladder and bowel management programs teach basic toileting routines, and patients also learn techniques for self-grooming. People acquire coping strategies for recurring episodes of spasticity, autonomic dysreflexia, and neurogenic pain. Vocational rehabilitation begins with an assessment of basic work skills, current dexterity, and physical and cognitive capabilities to determine the likelihood for employment. A vocational rehabilitation specialist then identifies potential work places, determines the type of assistive equipment that will be needed, and helps arrange for a user-friendly workplace. For those whose disabilities prevent them from returning to the workplace, therapists focus on encouraging productivity through participation in activities that provide a sense of satisfaction and self-esteem. This could include educational classes, hobbies, memberships in special interest groups, and participation in family and community events. Recreation therapy encourages patients to build on their abilities so that they can participate in recreational or athletic activities at their level of mobility. Engaging in recreational outlets and athletics helps those with spinal cord injuries achieve a more balanced and normal lifestyle and also provides opportunities for socialization and self-expression. How Is Research Helping Spinal Cord Injury Patients?Can an injured spinal cord be rebuilt? This is the question that drives basic research in the field of spinal cord injury. As investigators try to understand the underlying biological mechanisms that either inhibit or promote new growth in the spinal cord, they are making surprising discoveries, not just about how neurons and their axons grow in the CNS, but also about why they fail to regenerate after injury in the adult CNS. Understanding the cellular and molecular mechanisms involved in both the working and the damaged spinal cord could point the way to therapies that might prevent secondary damage, encourage axons to grow past injured areas, and reconnect vital neural circuits within the spinal cord and CNS. There has been successful research in a number of fields that may someday help people with spinal cord injuries. Genetic studies have revealed a number of molecules that encourage axon growth in the developing CNS but prevent it in the adult. Research into embryonic and adult stem cell biology has furthered knowledge about how cells communicate with each other. Basic research has helped describe the mechanisms involved in the mysterious process of apoptosis, in which large groups of seemingly healthy cells self-destruct. New rehabilitation therapies that retrain neural circuits through forced motion and electrical stimulation of muscle groups are helping injured patients regain lost function. Researchers, many of whom are supported by the National Institute of Neurological Disorders and Stroke (NINDS), are focused on advancing our understanding of the four key principles of spinal cord repair:
A spinal cord injury is complex. Repairing it has to take into account all of the different kinds of damage that occur during and after the injury. Because the molecular and cellular environment of the spinal cord is constantly changing from the moment of injury until several weeks or even months later, combination therapies will have to be designed to address specific types of damage at different points in time. Discoveries in Basic Research A decade ago, researchers demonstrated a small but significant neuroprotective and anti-inflammatory effect from an adrenal corticosteroid drug called methylprednisolone if it was given within 8 hours of injury. It is the only treatment currently available to limit the extent of spinal cord injury and its risks are relatively low. Researchers continue to search for additional anti-inflammatory treatments that might prove even more effective. Preliminary clinical trials of another compound, GM-1 ganglioside, indicate that it could be useful in preventing secondary damage in acute spinal cord injury. A large, randomized clinical trial suggested that it might also improve neurological recovery from spinal cord injury during rehabilitation. These observations and others have led to optimism that recovery can be improved by altering cellular responses immediately after injury. Using what they know about the mechanisms that cause secondary damage - excitotoxicity, inflammation, and cell suicide (apoptosis) - researchers are creating and testing additional neuroprotective therapies to prevent the spread of post-injury damage and preserve surrounding tissue. Some of the findings in these three different areas follow:
Discoveries in Clinical Research Advances in basic research are also being matched by progress in clinical research, especially in understanding the kinds of physical rehabilitation that work best to restore function. Some of the more promising rehabilitation techniques are helping spinal cord injury patients become more mobile.
The Future of Spinal Cord ResearchFueled by significant federal and private funding, the past decade of spinal cord injury research has produced a wealth of discoveries that are making the repair of injured spinal cords a reachable goal. This is good news for the 10,000 to 12,000 Americans every year who sustain these traumatic injuries. Because spinal cord injuries happen predominantly to people under the age of 30, the human cost is high. Major improvements in emergency and acute care have improved survival rates but have also increased the numbers of individuals who have to cope with severe disabilities for the rest of their lives. The cost to society, in terms of health care costs, disability payments, and lost income, is disproportionately high compared to other medical conditions. Considering the biological complexity of spinal cord injury, discovering successful ways to repair injuries and create rehabilitative strategies that significantly reduce disabilities is not an easy task. Researchers, many of them supported by the NINDS, are actively developing innovative research strategies aimed at making the kinds of exciting new discoveries that will translate into better clinical care and better lives for all. Where can I get more information? For more information on neurological disorders or research programs
funded by the National Institute of Neurological Disorders and
Stroke, contact the Institute's Brain Resources and Information
Network (BRAIN) at: BRAIN Information also is available from the following organizations:
agonist - a drug capable of combining with a receptor and initiating action. antagonist - a drug that opposes the effects of another by physiological or chemical action or by a competitive mechanism. apoptosis - also called programmed cell death. A form of cell death in which a programmed sequence of events leads to the elimination of old, unnecessary, and unhealthy cells. arrhythmia - an abnormal heart rhythm. The heartbeats may be too slow, too rapid, too irregular, or too early. astrocyte - a type of glial cell responsible for neurotransmission and neuronal metabolism. autonomic dysreflexia - a potentially dangerous complication of spinal cord injury in which blood pressure rises to dangerous levels. If not treated, autonomic dysreflexia can lead to stroke and possibly death. axial traction - the application of a mechanical force to stretch the spine; used to relieve pressure by separating vertebral surfaces and stretching soft tissues. axon - the long, thin extension of a nerve cell that conducts impulses away from the cell body. axonal growth cone - dynamic structures present at the tip of developing and regenerating axons that respond to chemical cues for growth and direction. central pattern generators (CPG) - neural circuits that produce self-sustaining patterns of behavior independent of their sensory input. Researchers have found evidence of a locomotor CPG in the spinal cord that synchronizes muscle activity during alternating stepping of the legs and feet. cervical - the part of the spine in the neck region. coccygeal - the part of the spine at the bottom of the spinal column, above the buttocks. cytokine - a small protein released by immune cells that has a specific effect on the interactions between cells, or communications between cells, or on the behavior of cells. dendrite - a short arm-like protuberance from a neuron. Dendrite is from the Greek for "branched like a tree." disc - shortened terminology for an intervertebral disc, a disc-shaped piece of specialized tissue that separates the bones of the spinal column. electroejaculation - a technique that uses an electric probe to stimulate ejaculation. embryonic stem cells - undifferentiated cells from the embryo that have the potential to become a wide variety of specialized cell types. excitotoxicity - a neurological process that is the result of the release of excessive amounts of the neurotransmitter glutamate. extracellular matrix - the material found around cells composed of structural proteins, specialized proteins, and proteoglycans. fetal spinal cord cells - cells used by scientists to derive undifferentiated embryonic stem cells for transplant into the damaged spinal cord. free radicals - highly reactive chemicals that attack molecules and modify their chemical structure. functional electrical stimulation (FES) - the therapeutic use of low-level electrical current to stimulate muscle movement and restore useful movements such as standing or stepping; also called functional neuromuscular stimulation. glia -supportive cells in the brain and spinal cord. Glial cells are the most abundant cell types in the central nervous system. There are three types: astrocytes, oligodendrocytes, and microglia. glutamate - an excitatory neurotransmitter. growth-inhibiting proteins: protein molecules that inhibit axon regeneration. guidance molecules - molecules that guide axons to their target. Some guidance molecules attract certain axons while repelling others. hypothermia - abnormally low body temperature. interneurons - neurons with axons that remain within the spinal cord. intubation - the process of putting a tube into a hollow organ or passageway, often into the airway. ligament - a tough band of connective tissue that connects various structures such as two bones. lumbar - the part of the spine in the middle back, below the thoracic vertebrae and above the sacral vertebrae. macrophage - a type of white blood cell that engulfs foreign material. Macrophages are key players in the immune response to foreign invaders such as infectious microorganisms Macrophages also release substances that stimulate other cells of the immune system. methylprednisolone - a steroid drug used to improve recovery from spinal cord injury. microglia - glial cells that function as part of the immune system in the brain and spinal cord. monocyte - a white blood cell that has a single nucleus and can engulf foreign material. Monocytes emigrate from blood into the tissues of the body and evolve into macrophages. myelin - a structure of cell membranes that forms a sheath around axons, insulating them and speeding conduction of nerve impulses. myelotomy - a surgical procedure that cuts into the spinal cord. neural prostheses - prosthetic devices that can respond to signals from the brain. neurogenic pain - generalized pain that results from nervous system malfunction. neuromodulation - a series of techniques employing electrical stimulation or the administration of medication by means of devices implanted in the body. These techniques allow the treatment of a range of disorders including certain forms of pain, spasticity, tremor, and urinary problems. neuron - also known as a nerve cell; the structural and functional unit of the nervous system. A neuron consists of a cell body and its processes: an axon and one or more dendrites. neurostimulation - the act of stimulating neurons with electrical impulses delivered via electrodes attached to the brain. neurotransmitter - a chemical released from neurons that transmits an impulse to another neuron, muscle, organ, or other tissue. neurotrophic factors - proteins responsible for the growth and survival of neurons. neutrophil - a type of white blood cell that engulfs, kills, and digests microorganisms. oligodendrocyte - a type of nerve cell in the brain and spinal cord that surrounds and insulates axons. olfactory ensheathing glia - non-myelinating glial cells that ensheath olfactory axons within both the PNS and CNS portions of the primary olfactory pathway. They are being used in experiments to build bridges between damaged areas of the spinal cord. paralysis - the inability to control movement of a part of the body. paraplegia - a condition involving complete paralysis of the legs. pressure sore (also known as a pressure ulcer or bed sore) - a reddened area or open sore caused by unrelieved pressure on the skin over bony areas such as the hip-bone or tailbone. quadriplegia - a condition involving complete paralysis of the legs and partial or complete paralysis of the arms. receptor - a structure on the surface or interior of a cell that selectively receives and binds to a specific substance. regeneration - repair, regrowth, or restoration of tissues; opposite of degeneration. rhizotomy - an operation to disconnect specific nerve roots in order to stop severe spasticity. sacral - refers to the part of the spine in the hip area. Schwann cell - the cell of the peripheral nervous system that forms the myelin sheath. spasticity - increased tone in muscles of the arms and legs (due to lesions of the upper motor neurons). spinal shock - a temporary physiological state that can occur after a spinal cord injury in which all sensory, motor, and sympathetic functions of the nervous system are lost below the level of injury. Spinal shock can lower blood pressure to dangerous levels and cause temporary paralysis. stem cell - special cells that have the ability to grow into any one of the body's more than 200 cell types. Unlike mature cells, which are permanently committed to their fate, stem cells can both renew themselves and create cells of other tissues. synapse - a specialized junction between two nerve cells. At the synapse, a neuron releases neurotransmitters that diffuse across the gap and activate receptors situated on the target cell. T-cell - an immune system cell that produces substances called cytokines, which stimulate the immune response. thoracic - the part of the spine at the upper-back to mid-back level. vertebrae - the 33 hollow bones that make up the spine. NIH Publication No. 03-160
|
Stroke: Hope Through Research
IntroductionMore than 2,400 years ago the father of medicine, Hippocrates, recognized and described stroke-the sudden onset of paralysis. Until recently, modern medicine has had very little power over this disease, but the world of stroke medicine is changing and new and better therapies are being developed every day. Today, some people who have a stroke can walk away from the attack with no or few disabilities if they are treated promptly. Doctors can finally offer stroke patients and their families the one thing that until now has been so hard to give: hope. In ancient times stroke was called apoplexy,* a general term that physicians applied to anyone suddenly struck down with paralysis. Because many conditions can lead to sudden paralysis, the term apoplexy did not indicate a specific diagnosis or cause. Physicians knew very little about the cause of stroke and the only established therapy was to feed and care for the patient until the attack ran its course. The first person to investigate the pathological signs of apoplexy was Johann Jacob Wepfer. Born in Schaffhausen, Switzerland, in 1620, Wepfer studied medicine and was the first to identify postmortem signs of bleeding in the brains of patients who died of apoplexy. From autopsy studies he gained knowledge of the carotid and vertebral arteries that supply the brain with blood. He also was the first person to suggest that apoplexy, in addition to being caused by bleeding in the brain, could be caused by a blockage of one of the main arteries supplying blood to the brain; thus stroke became known as a cerebrovascular disease ("cerebro" refers to a part of the brain; "vascular" refers to the blood vessels and arteries). Medical science would eventually confirm Wepfer's hypotheses, but until very recently doctors could offer little in the area of therapy. Over the last two decades basic and clinical investigators, many of them sponsored and funded in part by the National Institute of Neurological Disorders and Stroke (NINDS), have learned a great deal about stroke. They have identified major risk factors for the disease and have developed surgical techniques and drug treatments for the prevention of stroke. But perhaps the most exciting new development in the field of stroke research is the recent approval of a drug treatment that can reverse the course of stroke if given during the first few hours after the onset of symptoms. Studies with animals have shown that brain injury occurs within
minutes of a stroke and can become irreversible within as little
as an hour. In humans, brain damage begins from the moment the stroke
starts and often continues for days afterward. Scientists now know
that there is a very short window of opportunity for treatment of
the most common form of stroke. Because of these and other advances
in the field of cerebrovascular disease stroke patients now have
a chance for survival and recovery.
Cost of Stroke to the United States
* From "The Stroke/Brain Attack Reporter's Handbook," National Stroke Association, Englewood, CO, 1997 A stroke occurs when the blood supply to part of the brain is suddenly interrupted or when a blood vessel in the brain bursts, spilling blood into the spaces surrounding brain cells. In the same way that a person suffering a loss of blood flow to the heart is said to be having a heart attack, a person with a loss of blood flow to the brain or sudden bleeding in the brain can be said to be having a "brain attack." Brain cells die when they no longer receive oxygen and nutrients from the blood or when they are damaged by sudden bleeding into or around the brain. Ischemia is the term used to describe the loss of oxygen and nutrients for brain cells when there is inadequate blood flow. Ischemia ultimately leads to infarction, the death of brain cells which are eventually replaced by a fluid-filled cavity (or infarct) in the injured brain. When blood flow to the brain is interrupted, some brain cells die immediately, while others remain at risk for death. These damaged cells make up the ischemic penumbra and can linger in a compromised state for several hours. With timely treatment these cells can be saved. The ischemic penumbra is discussed in more detail in the Appendix. Even though a stroke occurs in the unseen reaches of the brain, the symptoms of a stroke are easy to spot. They include sudden numbness or weakness, especially on one side of the body; sudden confusion or trouble speaking or understanding speech; sudden trouble seeing in one or both eyes; sudden trouble walking, dizziness, or loss of balance or coordination; or sudden severe headache with no known cause. All of the symptoms of stroke appear suddenly, and often there is more than one symptom at the same time. Therefore stroke can usually be distinguished from other causes of dizziness or headache. These symptoms may indicate that a stroke has occurred and that medical attention is needed immediately. There are two forms of stroke: ischemic - blockage of a blood vessel supplying the brain, and hemorrhagic - bleeding into or around the brain. The following sections describe these forms in detail. An ischemic stroke occurs when an artery supplying the brain with blood becomes blocked, suddenly decreasing or stopping blood flow and ultimately causing a brain infarction. This type of stroke accounts for approximately 80 percent of all strokes. Blood clots are the most common cause of artery blockage and brain infarction. The process of clotting is necessary and beneficial throughout the body because it stops bleeding and allows repair of damaged areas of arteries or veins. However, when blood clots develop in the wrong place within an artery they can cause devastating injury by interfering with the normal flow of blood. Problems with clotting become more frequent as people age. Blood clots can cause ischemia and infarction in two ways. A clot that forms in a part of the body other than the brain can travel through blood vessels and become wedged in a brain artery. This free-roaming clot is called an embolus and often forms in the heart. A stroke caused by an embolus is called an embolic stroke. The second kind of ischemic stroke, called a thrombotic stroke, is caused by thrombosis, the formation of a blood clot in one of the cerebral arteries that stays attached to the artery wall until it grows large enough to block blood flow. Ischemic strokes can also be caused by stenosis, or a narrowing of the artery due to the buildup of plaque (a mixture of fatty substances, including cholesterol and other lipids) and blood clots along the artery wall. Stenosis can occur in large arteries and small arteries and is therefore called large vessel disease or small vessel disease, respectively. When a stroke occurs due to small vessel disease, a very small infarction results, sometimes called a lacunar infarction, from the French word "lacune" meaning "gap" or "cavity." The most common blood vessel disease that causes stenosis is atherosclerosis. In atherosclerosis, deposits of plaque build up along the inner walls of large and medium-sized arteries, causing thickening, hardening, and loss of elasticity of artery walls and decreased blood flow. The role of cholesterol and blood lipids with respect to stroke risk is discussed in the section on cholesterol under "Who is at Risk for Stroke?". In a healthy, functioning brain, neurons do not come into direct contact with blood. The vital oxygen and nutrients the neurons need from the blood come to the neurons across the thin walls of the cerebral capillaries. The glia (nervous system cells that support and protect neurons) form a blood-brain barrier, an elaborate meshwork that surrounds blood vessels and capillaries and regulates which elements of the blood can pass through to the neurons. When an artery in the brain bursts, blood spews out into the surrounding tissue and upsets not only the blood supply but the delicate chemical balance neurons require to function. This is called a hemorrhagic stroke. Such strokes account for approximately 20 percent of all strokes. Hemorrhage can occur in several ways. One common cause is a bleeding aneurysm, a weak or thin spot on an artery wall. Over time, these weak spots stretch or balloon out under high arterial pressure. The thin walls of these ballooning aneurysms can rupture and spill blood into the space surrounding brain cells. Hemorrhage also occurs when arterial walls break open. Plaque-encrusted artery walls eventually lose their elasticity and become brittle and thin, prone to cracking. Hypertension, or high blood pressure, increases the risk that a brittle artery wall will give way and release blood into the surrounding brain tissue. A person with an arteriovenous malformation (AVM) also has an increased risk of hemorrhagic stroke. AVMs are a tangle of defective blood vessels and capillaries within the brain that have thin walls and can therefore rupture. Bleeding from ruptured brain arteries can either go into the substance of the brain or into the various spaces surrounding the brain. Intracerebral hemorrhage occurs when a vessel within the brain leaks blood into the brain itself. Subarachnoid hemorrhage is bleeding under the meninges, or outer membranes, of the brain into the thin fluid-filled space that surrounds the brain. The subarachnoid space separates the arachnoid membrane from the underlying pia mater membrane. It contains a clear fluid (cerebrospinal fluid or CSF) as well as the small blood vessels that supply the outer surface of the brain. In a subarachnoid hemorrhage, one of the small arteries within the subarachnoid space bursts, flooding the area with blood and contaminating the cerebrospinal fluid. Since the CSF flows throughout the cranium, within the spaces of the brain, subarachnoid hemorrhage can lead to extensive damage throughout the brain. In fact, subarachnoid hemorrhage is the most deadly of all strokes. A transient ischemic attack (TIA), sometimes called a mini-stroke, starts just like a stroke but then resolves leaving no noticeable symptoms or deficits. The occurrence of a TIA is a warning that the person is at risk for a more serious and debilitating stroke. Of the approximately 50,000 Americans who have a TIA each year, about one-third will have an acute stroke sometime in the future. The addition of other risk factors compounds a person's risk for a recurrent stroke. The average duration of a TIA is a few minutes. For almost all TIAs, the symptoms go away within an hour. There is no way to tell whether symptoms will be just a TIA or persist and lead to death or disability. The patient should assume that all stroke symptoms signal an emergency and should not wait to see if they go away. Recurrent stroke is frequent; about 25 percent of people who recover from their first stroke will have another stroke within 5 years. Recurrent stroke is a major contributor to stroke disability and death, with the risk of severe disability or death from stroke increasing with each stroke recurrence. The risk of a recurrent stroke is greatest right after a stroke, with the risk decreasing with time. About 3 percent of stroke patients will have another stroke within 30 days of their first stroke and one-third of recurrent strokes take place within 2 years of the first stroke. Symptoms of stroke appear suddenly. Watch for these symptoms and be prepared to act quickly for yourself or on behalf of someone you are with:
If you suspect you or someone you know is experiencing any of these symptoms indicative of a stroke, do not wait. Call 911 emergency immediately. There are now effective therapies for stroke that must be administered at a hospital, but they lose their effectiveness if not given within the first 3 hours after stroke symptoms appear. Every minute counts! How is the Cause of Stroke Determined? Physicians have several diagnostic techniques and imaging tools to help diagnose the cause of stroke quickly and accurately. The first step in diagnosis is a short neurological examination. When a possible stroke patient arrives at a hospital, a health care professional, usually a doctor or nurse, will ask the patient or a companion what happened and when the symptoms began. Blood tests, an electrocardiogram, and a brain scan, such CT or MRI, will often be done. One test that helps doctors judge the severity of a stroke is the standardized NIH Stroke Scale, developed by the NINDS. Health care professionals use the NIH Stroke Scale to measure a patient's neurological deficits by asking the patient to answer questions and to perform several physical and mental tests. Other scales include the Glasgow Coma Scale, the Hunt and Hess Scale, the Modified Rankin Scale, and the Barthel Index. Imaging for the Diagnosis of Acute
Stroke Health care professionals also use a variety of imaging devices to evaluate stroke patients. The most widely used imaging procedure is the computed tomography (CT) scan. Also known as a CAT scan or computed axial tomography, CT creates a series of cross-sectional images of the head and brain. Because it is readily available at all hours at most major hospitals and produces images quickly, CT is the most commonly used diagnostic technique for acute stroke. CT also has unique diagnostic benefits. It will quickly rule out a hemorrhage, can occasionally show a tumor that might mimic a stroke, and may even show evidence of early infarction. Infarctions generally show up on a CT scan about 6 to 8 hours after the start of stroke symptoms. If a stroke is caused by hemorrhage, a CT can show evidence of bleeding into the brain almost immediately after stroke symptoms appear. Hemorrhage is the primary reason for avoiding certain drug treatments for stroke, such as thrombolytic therapy, the only proven acute stroke therapy for ischemic stroke (see section on "What Stroke Therapies are Available?"). Thrombolytic therapy cannot be used until the doctor can confidently diagnose the patient as suffering from an ischemic stroke because this treatment might increase bleeding and could make a hemorrhagic stroke worse. Another imaging device used for stroke patients is the magnetic resonance imaging (MRI) scan. MRI uses magnetic fields to detect subtle changes in brain tissue content. One effect of stroke is the slowing of water movement, called diffusion, through the damaged brain tissue. MRI can show this type of damage within the first hour after the stroke symptoms start. The benefit of MRI over a CT scan is more accurate and earlier diagnosis of infarction, especially for smaller strokes, while showing equivalent accuracy in determining when hemorrhage is present. MRI is more sensitive than CT for other types of brain disease, such as brain tumor, that might mimic a stroke. MRI cannot be performed in patients with certain types of metallic or electronic implants, such as pacemakers for the heart. Although increasingly used in the emergency diagnosis of stroke, MRI is not immediately available at all hours in most hospitals, where CT is used for acute stroke diagnosis. Also, MRI takes longer to perform than CT, and may not be performed if it would significantly delay treatment. Other types of MRI scans, often used for the diagnosis of cerebrovascular disease and to predict the risk of stroke, are magnetic resonance angiography (MRA) and functional magnetic resonance imaging (fMRI). Neurosurgeons use MRA to detect stenosis (blockage) of the brain arteries inside the skull by mapping flowing blood. Functional MRI uses a magnet to pick up signals from oxygenated blood and can show brain activity through increases in local blood flow. Duplex Doppler ultrasound and arteriography are two diagnostic imaging techniques used to decide if an individual would benefit from a surgical procedure called carotid endarterectomy. This surgery is used to remove fatty deposits from the carotid arteries and can help prevent stroke (see information on carotid endarterectomy). Doppler ultrasound is a painless, noninvasive test in which sound waves above the range of human hearing are sent into the neck. Echoes bounce off the moving blood and the tissue in the artery and can be formed into an image. Ultrasound is fast, painless, risk-free, and relatively inexpensive compared to MRA and arteriography, but it is not considered to be as accurate as arteriography. Arteriography is an X-ray of the carotid artery taken when a special dye is injected into the artery. The procedure carries its own small risk of causing a stroke and is costly to perform. The benefits of arteriography over MR techniques and ultrasound are that it is extremely reliable and still the best way to measure stenosis of the carotid arteries. Even so, significant advances are being made every day involving noninvasive imaging techniques such as fMRI (see section on surgery in "What Stroke Therapies are Available?"). Some people are at a higher risk for stroke than others. Unmodifiable risk factors include age, gender, race/ethnicity, and stroke family history. In contrast, other risk factors for stroke, like high blood pressure or cigarette smoking, can be changed or controlled by the person at risk. It is a myth that stroke occurs only in elderly adults. In actuality, stroke strikes all age groups, from fetuses still in the womb to centenarians. It is true, however, that older people have a higher risk for stroke than the general population and that the risk for stroke increases with age. For every decade after the age of 55, the risk of stroke doubles, and two-thirds of all strokes occur in people over 65 years old. People over 65 also have a seven-fold greater risk of dying from stroke than the general population. And the incidence of stroke is increasing proportionately with the increase in the elderly population. When the baby boomers move into the over-65 age group, stroke and other diseases will take on even greater significance in the health care field. Gender also plays a role in risk for stroke. Men have a higher risk for stroke, but more women die from stroke. The stroke risk for men is 1.25 times that for women. But men do not live as long as women, so men are usually younger when they have their strokes and therefore have a higher rate of survival than women. In other words, even though women have fewer strokes than men, women are generally older when they have their strokes and are more likely to die from them. Stroke seems to run in some families. Several factors might contribute to familial stroke risk. Members of a family might have a genetic tendency for stroke risk factors, such as an inherited predisposition for hypertension or diabetes. The influence of a common lifestyle among family members could also contribute to familial stroke. The risk for stroke varies among different ethnic and racial groups. The incidence of stroke among African-Americans is almost double that of white Americans, and twice as many African-Americans who have a stroke die from the event compared to white Americans. African-Americans between the ages of 45 and 55 have four to five times the stroke death rate of whites. After age 55 the stroke mortality rate for whites increases and is equal to that of African-Americans. Compared to white Americans, African-Americans have a higher incidence of stroke risk factors, including high blood pressure and cigarette smoking. African-Americans also have a higher incidence and prevalence of some genetic diseases, such as diabetes and sickle cell anemia, that predispose them to stroke. Hispanics and Native Americans have stroke incidence and mortality rates more similar to those of white Americans. In Asian-Americans stroke incidence and mortality rates are also similar to those in white Americans, even though Asians in Japan, China, and other countries of the Far East have significantly higher stroke incidence and mortality rates than white Americans. This suggests that environment and lifestyle factors play a large role in stroke risk. Several decades ago, scientists and statisticians noticed that people in the southeastern United States had the highest stroke mortality rate in the country. They named this region the stroke belt. For many years, researchers believed that the increased risk was due to the higher percentage of African-Americans and an overall lower socioeconomic status (SES) in the southern states. A low SES is associated with an overall lower standard of living, leading to a lower standard of health care and therefore an increased risk of stroke. But researchers now know that the higher percentage of African-Americans and the overall lower SES in the southern states does not adequately account for the higher incidence of, and mortality from, stroke in those states. This means that other factors must be contributing to the higher incidence of and mortality from stroke in this region. Recent studies have also shown that there is a stroke buckle in the stroke belt. Three southeastern states, North Carolina, South Carolina, and Georgia, have an extremely high stroke mortality rate, higher than the rate in other stroke belt states and up to two times the stroke mortality rate of the United States overall. The increased risk could be due to geographic or environmental factors or to regional differences in lifestyle, including higher rates of cigarette smoking and a regional preference for salty, high-fat foods. The most important risk factors for stroke are hypertension, heart disease, diabetes, and cigarette smoking. Others include heavy alcohol consumption, high blood cholesterol levels, illicit drug use, and genetic or congenital conditions, particularly vascular abnormalities. People with more than one risk factor have what is called "amplification of risk." This means that the multiple risk factors compound their destructive effects and create an overall risk greater than the simple cumulative effect of the individual risk factors. Of all the risk factors that contribute to stroke, the most powerful is hypertension, or high blood pressure. People with hypertension have a risk for stroke that is four to six times higher than the risk for those without hypertension. One-third of the adult U.S. population, about 50 million people (including 40-70 percent of those over age 65) have high blood pressure. Forty to 90 percent of stroke patients have high blood pressure before their stroke event. A systolic pressure of 120 mm of Hg over a diastolic pressure of 80 mm of Hg* is generally considered normal. Persistently high blood pressure greater than 140 over 90 leads to the diagnosis of the disease called hypertension. The impact of hypertension on the total risk for stroke decreases with increasing age, therefore factors other than hypertension play a greater role in the overall stroke risk in elderly adults. For people without hypertension, the absolute risk of stroke increases over time until around the age of 90, when the absolute risk becomes the same as that for people with hypertension. Like stroke, there is a gender difference in the prevalence of hypertension. In younger people, hypertension is more common among men than among women. With increasing age, however, more women than men have hypertension. This hypertension gender-age difference probably has an impact on the incidence and prevalence of stroke in these populations. Antihypertensive medication can decrease a person's risk for stroke. Recent studies suggest that treatment can decrease the stroke incidence rate by 38 percent and decrease the stroke fatality rate by 40 percent. Common hypertensive agents include adrenergic agents, beta-blockers, angiotensin converting enzyme inhibitors, calcium channel blockers, diuretics, and vasodilators. After hypertension, the second most powerful risk factor for stroke is heart disease, especially a condition known as atrial fibrillation. Atrial fibrillation is irregular beating of the left atrium, or left upper chamber, of the heart. In people with atrial fibrillation, the left atrium beats up to four times faster than the rest of the heart. This leads to an irregular flow of blood and the occasional formation of blood clots that can leave the heart and travel to the brain, causing a stroke. Atrial fibrillation, which affects as many as 2.2 million Americans, increases an individual's risk of stroke by 4 to 6 percent, and about 15 percent of stroke patients have atrial fibrillation before they experience a stroke. The condition is more prevalent in the upper age groups, which means that the prevalence of atrial fibrillation in the United States will increase proportionately with the growth of the elderly population. Unlike hypertension and other risk factors that have a lesser impact on the ever-rising absolute risk of stroke that comes with advancing age, the influence of atrial fibrillation on total risk for stroke increases powerfully with age. In people over 80 years old, atrial fibrillation is the direct cause of one in four strokes. Other forms of heart disease that increase stroke risk include malformations of the heart valves or the heart muscle. Some valve diseases, like mitral valve stenosis or mitral annular calcification, can double the risk for stroke, independent of other risk factors. Heart muscle malformations can also increase the risk for stroke. Patent foramen ovale (PFO) is a passage or a hole (sometimes called a "shunt") in the heart wall separating the two atria, or upper chambers, of the heart. Clots in the blood are usually filtered out by the lungs, but PFO could allow emboli or blood clots to bypass the lungs and go directly through the arteries to the brain, potentially causing a stroke. Research is currently under way to determine how important PFO is as a cause for stroke. Atrial septal aneurysm (ASA), a congenital (present from birth) malformation of the heart tissue, is a bulging of the septum or heart wall into one of the atria of the heart. Researchers do not know why this malformation increases the risk for stroke. PFO and ASA frequently occur together and therefore amplify the risk for stroke. Two other heart malformations that seem to increase the risk for stroke for unknown reasons are left atrial enlargement and left ventricular hypertrophy. People with left atrial enlargement have a larger than normal left atrium of the heart; those with left ventricular hypertrophy have a thickening of the wall of the left ventricle. Another risk factor for stroke is cardiac surgery to correct heart malformations or reverse the effects of heart disease. Strokes occurring in this situation are usually the result of surgically dislodged plaques from the aorta that travel through the bloodstream to the arteries in the neck and head, causing stroke. Cardiac surgery increases a person's risk of stroke by about 1 percent. Other types of surgery can also increase the risk of stroke. Most people know that high cholesterol levels contribute to heart disease. But many don't realize that a high cholesterol level also contributes to stroke risk. Cholesterol, a waxy substance produced by the liver, is a vital body product. It contributes to the production of hormones and vitamin D and is an integral component of cell membranes. The liver makes enough cholesterol to fuel the body's needs and this natural production of cholesterol alone is not a large contributing factor to atherosclerosis, heart disease, and stroke. Research has shown that the danger from cholesterol comes from a dietary intake of foods that contain high levels of cholesterol. Foods high in saturated fat and cholesterol, like meats, eggs, and dairy products, can increase the amount of total cholesterol in the body to alarming levels, contributing to the risk of atherosclerosis and thickening of the arteries. Cholesterol is classified as a lipid, meaning that it is fat-soluble rather than water-soluble. Other lipids include fatty acids, glycerides, alcohol, waxes, steroids, and fat-soluble vitamins A, D, and E. Lipids and water, like oil and water, do not mix. Blood is a water-based liquid, therefore cholesterol does not mix with blood. In order to travel through the blood without clumping together, cholesterol needs to be covered by a layer of protein. The cholesterol and protein together are called a lipoprotein. There are two kinds of cholesterol, commonly called the "good" and the "bad." Good cholesterol is high-density lipoprotein, or HDL; bad cholesterol is low-density lipoprotein, or LDL. Together, these two forms of cholesterol make up a person's total serum cholesterol level. Most cholesterol tests measure the level of total cholesterol in the blood and don't distinguish between good and bad cholesterol. For these total serum cholesterol tests, a level of less than 200 mg/dL** is considered safe, while a level of more than 240 is considered dangerous and places a person at risk for heart disease and stroke. Most cholesterol in the body is in the form of LDL. LDLs circulate through the bloodstream, picking up excess cholesterol and depositing cholesterol where it is needed (for example, for the production and maintenance of cell membranes). But when too much cholesterol starts circulating in the blood, the body cannot handle the excessive LDLs, which build up along the inside of the arterial walls. The buildup of LDL coating on the inside of the artery walls hardens and turns into arterial plaque, leading to stenosis and atherosclerosis. This plaque blocks blood vessels and contributes to the formation of blood clots. A person's LDL level should be less than 130 mg/dL to be safe. LDL levels between 130 and 159 put a person at a slightly higher risk for atherosclerosis, heart disease, and stroke. A score over 160 puts a person at great risk for a heart attack or stroke. The other form of cholesterol, HDL, is beneficial and contributes to stroke prevention. HDL carries a small percentage of the cholesterol in the blood, but instead of depositing its cholesterol on the inside of artery walls, HDL returns to the liver to unload its cholesterol. The liver then eliminates the excess cholesterol by passing it along to the kidneys. Currently, any HDL score higher than 35 is considered desirable. Recent studies have shown that high levels of HDL are associated with a reduced risk for heart disease and stroke and that low levels (less than 35 mg/dL), even in people with normal levels of LDL, lead to an increased risk for heart disease and stroke. A person may lower his risk for atherosclerosis and stroke by improving his cholesterol levels. A healthy diet and regular exercise are the best ways to lower total cholesterol levels. In some cases, physicians may prescribe cholesterol-lowering medication, and recent studies have shown that the newest types of these drugs, called reductase inhibitors or statin drugs, significantly reduce the risk for stroke in most patients with high cholesterol. Scientists believe that statins may work by reducing the amount of bad cholesterol the body produces and by reducing the body's inflammatory immune reaction to cholesterol plaque associated with atherosclerosis and stroke. * mm of Hg-or millimeters
of mercury-is the standard means of expressing blood pressure,
which is measured using an instrument called a sphygmomanometer.
Using a stethoscope and a cuff that is wrapped around the
patient's upper arm, a health professional listens to the
sounds of blood rushing through an artery. The first sound
registered on the instrument gauge (which measures the pressure
of the blood in millimeters on a column of mercury) is called
the systolic pressure. This is the maximum pressure produced
as the left ventricle of the heart contracts and the blood
begins to flow through the artery. The second sound is the
diastolic pressure and is the lowest pressure in the artery
when the left ventricle is relaxing. return to "Hypertension" section Diabetes is another disease that increases a person's risk for stroke. People with diabetes have three times the risk of stroke compared to people without diabetes. The relative risk of stroke from diabetes is highest in the fifth and sixth decades of life and decreases after that. Like hypertension, the relative risk of stroke from diabetes is highest for men at an earlier age and highest for women at an older age. People with diabetes may also have other contributing risk factors that can amplify the overall risk for stroke. For example, the prevalence of hypertension is 40 percent higher in the diabetic population compared to the general population. Modifiable Lifestyle Risk Factors Cigarette smoking is the most powerful modifiable stroke risk factor. Smoking almost doubles a person's risk for ischemic stroke, independent of other risk factors, and it increases a person's risk for subarachnoid hemorrhage by up to 3.5 percent. Smoking is directly responsible for a greater percentage of the total number of strokes in young adults than in older adults. Risk factors other than smoking - like hypertension, heart disease, and diabetes - account for more of the total number of strokes in older adults. Heavy smokers are at greater risk for stroke than light smokers. The relative risk of stroke decreases immediately after quitting smoking, with a major reduction of risk seen after 2 to 4 years. Unfortunately, it may take several decades for a former smoker's risk to drop to the level of someone who never smoked. Smoking increases the risk of stroke by promoting atherosclerosis and increasing the levels of blood-clotting factors, such as fibrinogen. In addition to promoting conditions linked to stroke, smoking also increases the damage that results from stroke by weakening the endothelial wall of the cerebrovascular system. This leads to greater damage to the brain from events that occur in the secondary stage of stroke. (The secondary effects of stroke are discussed in greater detail in the Appendix.) High alcohol consumption is another modifiable risk factor for stroke. Generally, an increase in alcohol consumption leads to an increase in blood pressure. While scientists agree that heavy drinking is a risk for both hemorrhagic and ischemic stroke, in several research studies daily consumption of smaller amounts of alcohol has been found to provide a protective influence against ischemic stroke, perhaps because alcohol decreases the clotting ability of platelets in the blood. Moderate alcohol consumption may act in the same way as aspirin to decrease blood clotting and prevent ischemic stroke. Heavy alcohol consumption, though, may seriously deplete platelet numbers and compromise blood clotting and blood viscosity, leading to hemorrhage. In addition, heavy drinking or binge drinking can lead to a rebound effect after the alcohol is purged from the body. The consequences of this rebound effect are that blood viscosity (thickness) and platelet levels skyrocket after heavy drinking, increasing the risk for ischemic stroke. The use of illicit drugs, such as cocaine and crack cocaine, can cause stroke. Cocaine may act on other risk factors, such as hypertension, heart disease, and vascular disease, to trigger a stroke. It decreases relative cerebrovascular blood flow by up to 30 percent, causes vascular constriction, and inhibits vascular relaxation, leading to narrowing of the arteries. Cocaine also affects the heart, causing arrhythmias and rapid heart rate that can lead to the formation of blood clots. Marijuana smoking may also be a risk factor for stroke. Marijuana decreases blood pressure and may interact with other risk factors, such as hypertension and cigarette smoking, to cause rapidly fluctuating blood pressure levels, damaging blood vessels. Other drugs of abuse, such as amphetamines, heroin, and anabolic steroids (and even some common, legal drugs, such as caffeine and L-asparaginase and pseudoephedrine found in over-the-counter decongestants), have been suspected of increasing stroke risk. Many of these drugs are vasoconstrictors, meaning that they cause blood vessels to constrict and blood pressure to rise. Injuries to the head or neck may damage the cerebrovascular system and cause a small number of strokes. Head injury or traumatic brain injury may cause bleeding within the brain leading to damage akin to that caused by a hemorrhagic stroke. Neck injury, when associated with spontaneous tearing of the vertebral or carotid arteries caused by sudden and severe extension of the neck, neck rotation, or pressure on the artery, is a contributing cause of stroke, especially in young adults. This type of stroke is often called "beauty-parlor syndrome," which refers to the practice of extending the neck backwards over a sink for hair-washing in beauty parlors. Neck calisthenics, "bottoms-up" drinking, and improperly performed chiropractic manipulation of the neck can also put strain on the vertebral and carotid arteries, possibly leading to ischemic stroke. Recent viral and bacterial infections may act with other risk factors to add a small risk for stroke. The immune system responds to infection by increasing inflammation and increasing the infection-fighting properties of the blood. Unfortunately, this immune response increases the number of clotting factors in the blood, leading to an increased risk of embolic-ischemic stroke. Although there may not be a single genetic factor associated with stroke, genes do play a large role in the expression of stroke risk factors such as hypertension, heart disease, diabetes, and vascular malformations. It is also possible that an increased risk for stroke within a family is due to environmental factors, such as a common sedentary lifestyle or poor eating habits, rather than hereditary factors. Vascular malformations that cause stroke may have the strongest genetic link of all stroke risk factors. A vascular malformation is an abnormally formed blood vessel or group of blood vessels. One genetic vascular disease called CADASIL, which stands for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. CADASIL is a rare, genetically inherited, congenital vascular disease of the brain that causes strokes, subcortical dementia, migraine-like headaches, and psychiatric disturbances. CADASIL is very debilitating and symptoms usually surface around the age of 45. Although CADASIL can be treated with surgery to repair the defective blood vessels, patients often die by the age of 65. The exact incidence of CADASIL in the United States is unknown. Medication or drug therapy is the most common treatment for stroke. The most popular classes of drugs used to prevent or treat stroke are antithrombotics (antiplatelet agents and anticoagulants) and thrombolytics. Antithrombotics prevent the formation of blood clots that can become lodged in a cerebral artery and cause strokes. Antiplatelet drugs prevent clotting by decreasing the activity of platelets, blood cells that contribute to the clotting property of blood. These drugs reduce the risk of blood-clot formation, thus reducing the risk of ischemic stroke. In the context of stroke, physicians prescribe antiplatelet drugs mainly for prevention. The most widely known and used antiplatelet drug is aspirin. Other antiplatelet drugs include clopidogrel, ticlopidine, and dipyridamole. The NINDS sponsors a wide range of clinical trials to determine the effectiveness of antiplatelet drugs for stroke prevention. Anticoagulants reduce stroke risk by reducing the clotting property of the blood. The most commonly used anticoagulants include warfarin (also known as Coumadin® ), heparin, and enoxaparin (also known as Lovenox). The NINDS has sponsored several trials to test the efficacy of anticoagulants versus antiplatelet drugs. The Stroke Prevention in Atrial Fibrillation (SPAF) trial found that, although aspirin is an effective therapy for the prevention of a second stroke in most patients with atrial fibrillation, some patients with additional risk factors do better on warfarin therapy. Another study, the Trial of Org 10127 in Acute Stroke Treatment (TOAST), tested the effectiveness of low-molecular weight heparin (Org 10172) in stroke prevention. TOAST showed that heparin anticoagulants are not generally effective in preventing recurrent stroke or improving outcome. Thrombolytic agents are used to treat an ongoing, acute ischemic stroke caused by an artery blockage. These drugs halt the stroke by dissolving the blood clot that is blocking blood flow to the brain. Recombinant tissue plasminogen activator (rt-PA) is a genetically engineered form of t-PA, a thombolytic substance made naturally by the body. It can be effective if given intravenously within 3 hours of stroke symptom onset, but it should be used only after a physician has confirmed that the patient has suffered an ischemic stroke. Thrombolytic agents can increase bleeding and therefore must be used only after careful patient screening. The NINDS rt-PA Stroke Study showed the efficacy of t-PA and in 1996 led to the first FDA-approved treatment for acute ischemic stroke. Other thrombolytics are currently being tested in clinical trials. Neuroprotectants are medications that protect the brain from secondary injury caused by stroke (see Appendix). Although no neuroprotectants are FDA-approved for use in stroke at this time, many are in clinical trials. There are several different classes of neuroprotectants that show promise for future therapy, including glutamate antagonists, antioxidants, apoptosis inhibitors, and many others. Surgery can be used to prevent stroke, to treat acute stroke, or to repair vascular damage or malformations in and around the brain. There are two prominent types of surgery for stroke prevention and treatment: carotid endarterectomy and extracranial/intracranial (EC/IC) bypass. Carotid endarterectomy is a surgical procedure in which a doctor removes fatty deposits (plaque) from the inside of one of the carotid arteries, which are located in the neck and are the main suppliers of blood to the brain. As mentioned earlier, the disease atherosclerosis is characterized by the buildup of plaque on the inside of large arteries, and the blockage of an artery by this fatty material is called stenosis. The NINDS has sponsored two large clinical trials to test the efficacy of carotid endarterectomy: the North American Symptomatic Carotid Endarterectomy Trial (NASCET) and the Asymptomatic Carotid Atherosclerosis Trial (ACAS). These trials showed that carotid endarterectomy is a safe and effective stroke prevention therapy for most people with greater than 50 percent stenosis of the carotid arteries when performed by a qualified and experienced neurosurgeon or vascular surgeon. Currently, the NINDS is sponsoring the Carotid Revascularization Endarterectomy vs. Stenting Trial (CREST), a large clinical trial designed to test the effectiveness of carotid endarterectomy versus a newer surgical procedure for carotid stenosis called stenting. The procedure involves inserting a long, thin catheter tube into an artery in the leg and threading the catheter through the vascular system into the narrow stenosis of the carotid artery in the neck. Once the catheter is in place in the carotid artery, the radiologist expands the stent with a balloon on the tip of the catheter. The CREST trial will test the effectiveness of the new surgical technique versus the established standard technique of carotid endarterectomy surgery. EC/IC bypass surgery is a procedure that restores blood flow to a blood-deprived area of brain tissue by rerouting a healthy artery in the scalp to the area of brain tissue affected by a blocked artery. The NINDS-sponsored EC/IC Bypass Study tested the ability of this surgery to prevent recurrent strokes in stroke patients with atherosclerosis. The study showed that, in the long run, EC/IC does not seem to benefit these patients. The surgery is still performed occasionally for patients with aneurysms, some types of small artery disease, and certain vascular abnormalities. One useful surgical procedure for treatment of brain aneurysms that cause subarachnoid hemorrhage is a technique called "clipping." Clipping involves clamping off the aneurysm from the blood vessel, which reduces the chance that it will burst and bleed. A new therapy that is gaining wide attention is the detachable coil technique for the treatment of high-risk intracranial aneurysms. A small platinum coil is inserted through an artery in the thigh and threaded through the arteries to the site of the aneurysm. The coil is then released into the aneurysm, where it evokes an immune response from the body. The body produces a blood clot inside the aneurysm, strengthening the artery walls and reducing the risk of rupture. Once the aneurysm is stabilized, a neurosurgeon can clip the aneurysm with less risk of hemorrhage and death to the patient.
Stroke is the number one cause of serious adult disability in the United States. Stroke disability is devastating to the stroke patient and family, but therapies are available to help rehabilitate post-stroke patients. For most stroke patients, physical therapy (PT) is the cornerstone of the rehabilitation process. A physical therapist uses training, exercises, and physical manipulation of the stroke patient's body with the intent of restoring movement, balance, and coordination. The aim of PT is to have the stroke patient relearn simple motor activities such as walking, sitting, standing, lying down, and the process of switching from one type of movement to another. Another type of therapy involving relearning daily activities is occupational therapy (OT). OT also involves exercise and training to help the stroke patient relearn everyday activities such as eating, drinking, dressing, bathing, cooking, reading and writing, and toileting. The goal of OT is to help the patient become independent or semi-independent. Speech and language problems arise when brain damage occurs in the language centers of the brain. Due to the brain's great ability to learn and change (called brain plasticity), other areas can adapt to take over some of the lost functions. Speech language pathologists help stroke patients relearn language and speaking skills, including swallowing, or learn other forms of communication. Speech therapy is appropriate for any patients with problems understanding speech or written words, or problems forming speech. A speech therapist helps stroke patients help themselves by working to improve language skills, develop alternative ways of communicating, and develop coping skills to deal with the frustration of not being able to communicate fully. With time and patience, a stroke survivor should be able to regain some, and sometimes all, language and speaking abilities. Many stroke patients require psychological or psychiatric help after a stroke. Psychological problems, such as depression, anxiety, frustration, and anger, are common post-stroke disabilities. Talk therapy, along with appropriate medication, can help alleviate some of the mental and emotional problems that result from stroke. Sometimes it is also beneficial for family members of the stroke patient to seek psychological help as well. For more information on rehabilitation, contact the National Rehabilitation Information Center , a service of the National Institute on Disability and Rehabilitation Research (see Where I can get more information). What Disabilities Can Result From
a Stroke? Although stroke is a disease of the brain, it can affect the entire body. Some of the disabilities that can result from a stroke include paralysis, cognitive deficits, speech problems, emotional difficulties, daily living problems, and pain. Paralysis: A common disability that results from stroke is complete paralysis on one side of the body, called hemiplegia. A related disability that is not as debilitating as paralysis is one-sided weakness or hemiparesis. The paralysis or weakness may affect only the face, an arm, or a leg or may affect one entire side of the body and face. A person who suffers a stroke in the left hemisphere of the brain will show right-sided paralysis or paresis. Conversely, a person with a stroke in the right hemisphere of the brain will show deficits on the left side of the body. A stroke patient may have problems with the simplest of daily activities, such as walking, dressing, eating, and using the bathroom. Motor deficits can result from damage to the motor cortex in the frontal lobes of the brain or from damage to the lower parts of the brain, such as the cerebellum, which controls balance and coordination. Some stroke patients also have trouble swallowing, called dysphagia. Cognitive deficits: Stroke may cause problems with thinking, awareness, attention, learning, judgment, and memory. In some cases of stroke, the patient suffers a "neglect" syndrome. The neglect means that a stroke patient has no knowledge of one side of his or her body, or one side of the visual field, or is unaware of the deficit. A stroke patient may be unaware of his or her surroundings, or may be unaware of the mental deficits that resulted from the stroke. Language deficits: Stroke victims often have problems understanding or forming speech. A deficit in understanding or forming speech is called aphasia. Aphasia usually occurs along with similar problems in reading or writing. In most people, language problems result from damage to the left hemisphere of the brain. Slurred speech due to weakness or incoordination of the muscles involved in speaking is called dysarthria, and is not a problem with language. Because it can result from any weakness or incoordination of the speech muscles, dysarthria can arise from damage to either side of the brain. Emotional deficits: A stroke can lead to emotional problems. Stroke patients may have difficulty controlling their emotions or may express inappropriate emotions in certain situations. One common disability that occurs with many stroke patients is depression. Post-stroke depression may be more than a general sadness resulting from the stroke incident. It is a clinical behavioral problem that can hamper recovery and rehabilitation and may even lead to suicide. Post-stroke depression is treated as any depression is treated, with antidepressant medications and therapy. Pain: Stroke patients may experience pain, uncomfortable numbness, or strange sensations after a stroke. These sensations may be due to many factors including damage to the sensory regions of the brain, stiff joints, or a disabled limb. An uncommon type of pain resulting from stroke is called central stroke pain or central pain syndrome (CPS). CPS results from damage to an area in the mid-brain called the thalamus. The pain is a mixture of sensations, including heat and cold, burning, tingling, numbness, and sharp stabbing and underlying aching pain. The pain is often worse in the extremities - the hands and feet - and is made worse by movement and temperature changes, especially cold temperatures. Unfortunately, since most pain medications provide little relief from these sensations, very few treatments or therapies exist to combat CPS. What Special Risks do Women Face? Some risk factors for stroke apply only to women. Primary among these are pregnancy, childbirth, and menopause. These risk factors are tied to hormonal fluctuations and changes that affect a woman in different stages of life. Research in the past few decades has shown that high-dose oral contraceptives, the kind used in the 1960s and 1970s, can increase the risk of stroke in women. Fortunately, oral contraceptives with high doses of estrogen are no longer used and have been replaced with safer and more effective oral contraceptives with lower doses of estrogen. Some studies have shown the newer low-dose oral contraceptives may not significantly increase the risk of stroke in women. Other studies have demonstrated that pregnancy and childbirth can put a woman at an increased risk for stroke. Pregnancy increases the risk of stroke as much as three to 13 times. Of course, the risk of stroke in young women of childbearing years is very small to begin with, so a moderate increase in risk during pregnancy is still a relatively small risk. Pregnancy and childbirth cause strokes in approximately eight in 100,000 women. Unfortunately, 25 percent of strokes during pregnancy end in death, and hemorrhagic strokes, although rare, are still the leading cause of maternal death in the United States. Subarachnoid hemorrhage, in particular, causes one to five maternal deaths per 10,000 pregnancies. A study sponsored by the NINDS showed that the risk of stroke during pregnancy is greatest in the post-partum period - the 6 weeks following childbirth. The risk of ischemic stroke after pregnancy is about nine times higher and the risk of hemorrhagic stroke is more than 28 times higher for post-partum women than for women who are not pregnant or post-partum. The cause is unknown. In the same way that the hormonal changes during pregnancy and childbirth are associated with increased risk of stroke, hormonal changes at the end of the childbearing years can increase the risk of stroke. Several studies have shown that menopause, the end of a woman's reproductive ability marked by the termination of her menstrual cycle, can increase a woman's risk of stroke. Fortunately, some studies have suggested that hormone replacement therapy can reduce some of the effects of menopause and decrease stroke risk. Currently, the NINDS is sponsoring the Women's Estrogen for Stroke Trial (WEST), a randomized, placebo-controlled, double-blind trial, to determine whether estrogen therapy can reduce the risk of death or recurrent stroke in postmenopausal women who have a history of a recent TIA or non-disabling stroke. The mechanism by which estrogen can prove beneficial to postmenopausal women could include its role in cholesterol control. Studies have shown that estrogen acts to increase levels of HDL while decreasing LDL levels. Are Children at Risk For Stroke? The young have several risk factors unique to them. Young people seem to suffer from hemorrhagic strokes more than ischemic strokes, a significant difference from older age groups where ischemic strokes make up the majority of stroke cases. Hemorrhagic strokes represent 20 percent of all strokes in the United States and young people account for many of these. Clinicians often separate the "young" into two categories: those younger than 15 years of age, and those 15 to 44 years of age. People 15 to 44 years of age are generally considered young adults and have many of the risk factors mentioned above, such as drug use, alcohol abuse, pregnancy, head and neck injuries, heart disease or heart malformations, and infections. Some other causes of stroke in the young are linked to genetic diseases. Medical complications that can lead to stroke in children include intracranial infection, brain injury, vascular malformations such as moyamoya syndrome, occlusive vascular disease, and genetic disorders such as sickle cell anemia, tuberous sclerosis, and Marfan's syndrome. The symptoms of stroke in children are different from those in adults and young adults. A child experiencing a stroke may have seizures, a sudden loss of speech, a loss of expressive language (including body language and gestures), hemiparesis (weakness on one side of the body), hemiplegia (paralysis on one side of the body), dysarthria (impairment of speech), convulsions, headache, or fever. It is a medical emergency when a child shows any of these symptoms. In children with stroke the underlying conditions that led to the stroke should be determined and managed to prevent future strokes. For example, a recent clinical study sponsored by the National Heart, Lung, and Blood Institute found that giving blood transfusions to young children with sickle cell anemia greatly reduces the risk of stroke. The Institute even suggests attempting to prevent stroke in high-risk children by giving them blood transfusions before they experience a stroke. Most children who experience a stroke will do better than most adults after treatment and rehabilitation. This is due in part to the immature brain's great plasticity, the ability to adapt to deficits and injury. Children who experience seizures along with stroke do not recover as well as children who do not have seizures. Some children may experience residual hemiplegia, though most will eventually learn how to walk. What Research is Being Done by the
NINDS? The NINDS is the leading supporter of stroke research in the United States and sponsors a wide range of experimental research studies, from investigations of basic biological mechanisms to studies with animal models and clinical trials. Currently, NINDS researchers are studying the mechanisms of stroke risk factors and the process of brain damage that results from stroke. Some of this brain damage may be secondary to the initial death of brain cells caused by the lack of blood flow to the brain tissue. This secondary wave of brain injury is a result of a toxic reaction to the primary damage and mainly involves the excitatory neurochemical, glutamate. Glutamate in the normal brain functions as a chemical messenger between brain cells, allowing them to communicate. But an excess amount of glutamate in the brain causes too much activity and brain cells quickly "burn out" from too much excitement, releasing more toxic chemicals, such as caspases, cytokines, monocytes, and oxygen-free radicals. These substances poison the chemical environment of surrounding cells, initiating a cascade of degeneration and programmed cell death, called apoptosis. NINDS researchers are studying the mechanisms underlying this secondary insult, which consists mainly of inflammation, toxicity, and a breakdown of the blood vessels that provide blood to the brain. Researchers are also looking for ways to prevent secondary injury to the brain by providing different types of neuroprotection for salvagable cells that prevent inflammation and block some of the toxic chemicals created by dying brain cells. From this research, scientists hope to develop neuroprotective agents to prevent secondary damage. For more information on excitotoxicity, neuroprotection, and the ischemic cascade, please refer to the Appendix. Basic research has also focused on the genetics of stroke and stroke risk factors. One area of research involving genetics is gene therapy. Gene therapy involves putting a gene for a desired protein in certain cells of the body. The inserted gene will then "program" the cell to produce the desired protein. If enough cells in the right areas produce enough protein, then the protein could be therapeutic. Scientists must find ways to deliver the therapeutic DNA to the appropriate cells and must learn how to deliver enough DNA to enough cells so that the tissues produce a therapeutic amount of protein. Gene therapy is in the very early stages of development and there are many problems to overcome, including learning how to penetrate the highly impermeable blood-brain barrier and how to halt the host's immune reaction to the virus that carries the gene to the cells. Some of the proteins used for stroke therapy could include neuroprotective proteins, anti-inflammatory proteins, and DNA/cellular repair proteins, among others. The NINDS supports and conducts a wide variety of studies in animals, from genetics research on zebrafish to rehabilitation research on primates. Much of the Institute's animal research involves rodents, specifically mice and rats. For example, one study of hypertension and stroke uses rats that have been bred to be hypertensive and therefore stroke-prone. By studying stroke in rats, scientists hope to get a better picture of what might be happening in human stroke patients. Scientists can also use animal models to test promising therapeutic interventions for stroke. If a therapy proves to be beneficial to animals, then scientists can consider testing the therapy in human subjects. One promising area of stroke animal research involves hibernation. The dramatic decrease of blood flow to the brain in hibernating animals is extensive - extensive enough that it would kill a non-hibernating animal. During hibernation, an animal's metabolism slows down, body temperature drops, and energy and oxygen requirements of brain cells decrease. If scientists can discover how animals hibernate without experiencing brain damage, then maybe they can discover ways to stop the brain damage associated with decreased blood flow in stroke patients. Other studies are looking at the role of hypothermia, or decreased body temperature, on metabolism and neuroprotection. Both hibernation and hypothermia have a relationship to hypoxia and edema. Hypoxia, or anoxia, occurs when there is not enough oxygen available for brain cells to function properly. Since brain cells require large amounts of oxygen for energy requirements, they are especially vulnerable to hypoxia. Edema occurs when the chemical balance of brain tissue is disturbed and water or fluids flow into the brain cells, making them swell and burst, releasing their toxic contents into the surrounding tissues. Edema is one cause of general brain tissue swelling and contributes to the secondary injury associated with stroke. The basic and animal studies discussed above do not involve people and fall under the category of preclinical research; clinical research involves people. One area of investigation that has made the transition from animal models to clinical research is the study of the mechanisms underlying brain plasticity and the neuronal rewiring that occurs after a stroke. New advances in imaging and rehabilitation have shown that the brain can compensate for function lost as a result of stroke. When cells in an area of the brain responsible for a particular function die after a stroke, the patient becomes unable to perform that function. For example, a stroke patient with an infarct in the area of the brain responsible for facial recognition becomes unable to recognize faces, a syndrome called facial agnosia. But, in time, the person may come to recognize faces again, even though the area of the brain originally programmed to perform that function remains dead. The plasticity of the brain and the rewiring of the neural connections make it possible for one part of the brain to change functions and take up the more important functions of a disabled part. This rewiring of the brain and restoration of function, which the brain tries to do automatically, can be helped with therapy. Scientists are working to develop new and better ways to help the brain repair itself to restore important functions to the stroke patient. One example of a therapy resulting from this research is the use of transcranial magnetic stimulation (TMS) in stroke rehabilitation. Some evidence suggests that TMS, in which a small magnetic current is delivered to an area of the brain, may possibly increase brain plasticity and speed up recovery of function after a stroke. The TMS device is a small coil which is held outside of the head, over the part of the brain needing stimulation. Currently, several studies at the NINDS are testing whether TMS has any value in increasing motor function and improving functional recovery. Clinical research is usually conducted in a series of trials that become progressively larger. A phase I clinical trial is directly built upon the lessons learned from basic and animal research and is used to test the safety of therapy for a particular disease and to estimate possible efficacy in a few human subjects. A phase II clinical trial usually involves many subjects at several different centers and is used to test safety and possible efficacy on a broader scale, to test different dosing for medications or to perfect techniques for surgery, and to determine the best methodology and outcome measures for the bigger phase III clinical trial to come. A phase III clinical trial is the largest endeavor in clinical research. This type of trial often involves many centers and many subjects. The trial usually has two patient groups who receive different treatments, but all other standard care is the same and represents the best care available. The trial may compare two treatments, or, if there is only one treatment to test, patients who do not receive the test therapy receive instead a placebo. The patients are told that the additional treatment they are receiving may be either the active treatment or a placebo. Many phase III trials are called double-blind, randomized clinical trials. Double-blind means that neither the subjects nor the doctors and nurses who are treating the subjects and determining the response to the therapy know which treatment a subject receives. Randomization refers to the placing of subjects into one of the treatment groups in a way that can't be predicted by the patients or investigators. These clinical trials usually involve many investigators and take many years to complete. The hypothesis and methods of the trial are very precise and well thought out. Clinical trial designs, as well as the concepts of blinding and randomization, have developed over years of experimentation, trial, and error. At the present time, researchers are developing new designs to maximize the opportunity for all subjects to receive therapy. Most treatments for general use come out of phase III clinical trials. After one or more phase III trials are finished, and if the results are positive for the treatment, the investigators can petition the FDA for government approval to use the drug or procedure to treat patients. Once the treatment is approved by the FDA, it can be used by qualified doctors throughout the country. The back packet of this brochure contains cards with information on some of the many stroke clinical trials the NINDS supports or has completed. NINDS-Sponsored Stroke Clinical
Trials: April 2007 Clinical trials give researchers a way to test new treatments in human subjects. Clinical trials test surgical devices and procedures, medications, rehabilitation therapies, and lifestyle and psychosocial interventions to determine how safe and effective they are and to establish the proper amount or level of treatment. Because of their scope and the need for careful analysis of data and outcomes, clinical trials are usually conducted in three phases and can take several years or more to complete.
NINDS conducts clinical trials at the NIH Clinical Center and also provides funding for clinical trials at hospitals and universities across the United States and Canada. Below are findings from some of the largest and most significant recent clinical trials in stroke, as well as summaries of some of the most promising clinical trials in progress. Findings From Recently Completed
Clinical Trials Warfarin vs. Aspirin for Intracranial Arterial Stenosis
(WASID) Extremity Constraint-Induced Therapy Evaluation (EXCITE) Preliminary Data from Ongoing Trials The Carotid Revascularization Endarterectomy vs. Stenting
Trial (CREST) Carotid Occlusion Surgery Study (COSS) Warfarin vs. Aspirin in Reduced Cardiac Ejection Fraction
(WARCEF) Secondary Prevention of Small Subcortical Strokes
(SPS3) Field Administration of Stroke Therapy Magnesium Trial
(FAST-MAG) Insulin Resistance Intervention After Stroke Trial
(IRIS) Interventional Management of Stroke Trial (IMS III) A Randomized Trial of Unruptured Brain Arteriovenous
Malformations (ARUBA) Where can I get more information? For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at: BRAIN Information also is available from the following organizations:
What Stroke Therapies are Available? Physicians have a wide range of therapies to choose from when determining a stroke patient's best therapeutic plan. The type of stroke therapy a patient should receive depends upon the stage of disease. Generally there are three treatment stages for stroke: prevention, therapy immediately after stroke, and post-stroke rehabilitation. Therapies to prevent a first or recurrent stroke are based on treating an individual's underlying risk factors for stroke, such as hypertension, atrial fibrillation, and diabetes, or preventing the widespread formation of blood clots that can cause ischemic stroke in everyone, whether or not risk factors are present. Acute stroke therapies try to stop a stroke while it is happening by quickly dissolving a blood clot causing the stroke or by stopping the bleeding of a hemorrhagic stroke. The purpose of post-stroke rehabilitation is to overcome disabilities that result from stroke damage. Therapies for stroke include medications, surgery, or rehabilitation. acute stroke-a stage of stroke starting at the onset of symptoms and last for a few hours thereafter. agnosia-a cognitive disability characterized by ignorance of or inability to acknowledge one side of the body or one side of the visual field. aneurysm -a weak or thin spot on an artery wall that has stretched or ballooned out from the wall and filled with blood, or damage to an artery leading to pooling of blood between the layers of the blood vessel walls. anoxia-a state of almost no oxygen delivery to a cell, resulting in low energy production and possible death of the cell; see hypoxia. anticoagulants-a drug therapy used to prevent the formation of blood clots that can become lodged in cerebral arteries and cause strokes. antiplatelet agents-a type of anticoagulant drug therapy that prevents the formation of blood clots by preventing the accumulation of platelets that form the basis of blood clots; some common antiplatelets include aspirin and ticlopidine; see anticoagulants. antithrombotics-a type of anticoagulant drug therapy that prevents the formation of blood clots by inhibiting the coagulating actions of the blood protein thrombin; some common antithrombotics include warfarin and heparin; see anticoagulants. aphasia-the inability to understand or create speech, writing, or language in general due to damage to the speech centers of the brain. apoplexy-a historical, but obsolete term for a cerebral stroke, most often intracerebral hemorrhage, that was applied to any condition that involved disorientation and/or paralysis. apoptosis- a form of cell death involving shrinking of the cell and eventual disposal of the internal elements of the cell by the body's immune system. Apoptosis is an active, non-toxic form of cell suicide that does not induce an inflammatory response. It is often called programmed cell death because it is triggered by a genetic signal, involves specific cell mechanisms, and is irreversible once initiated. apraxia-a movement disorder characterized by the inability to perform skilled or purposeful voluntary movements, generally caused by damage to the areas of the brain responsible for voluntary movement. arteriography-an X-ray of the carotid artery taken when a special dye is injected into the artery. arteriovenous malformation (AVM)-a congenital disorder characterized by a complex tangled web of arteries and veins. atherosclerosis-a blood vessel disease characterized by deposits of lipid material on the inside of the walls of large to medium-sized arteries which make the artery walls thick, hard, brittle, and prone to breaking. atrial fibrillation-irregular beating of the left atrium, or left upper chamber, of the heart. blood-brain barrier-an elaborate network of supportive brain cells, called glia, that surrounds blood vessels and protects neurons from the toxic effects of direct exposure to blood. carotid artery-an artery, located on either side of the neck, that supplies the brain with blood. carotid endarterectomy-surgery used to remove fatty deposits from the carotid arteries. central stroke pain (central pain syndrome)-pain caused by damage to an area in the thalamus. The pain is a mixture of sensations, including heat and cold, burning, tingling, numbness, and sharp stabbing and underlying aching pain. cerebral blood flow (CBF)-the flow of blood through the arteries that lead to the brain, called the cerebrovascular system. cerebrospinal fluid (CSF)-clear fluid that bathes the brain and spinal cord. cerebrovascular disease-a reduction in the supply of blood to the brain either by narrowing of the arteries through the buildup of plaque on the inside walls of the arteries, called stenosis, or through blockage of an artery due to a blood clot. cholesterol-a waxy substance, produced naturally by the liver and also found in foods, that circulates in the blood and helps maintain tissues and cell membranes. Excess cholesterol in the body can contribute to atherosclerosis and high blood pressure. "clipping"-surgical procedure for treatment of brain aneurysms, involving clamping an aneurysm from a blood vessel, surgically removing this ballooned part of the blood vessel, and closing the opening in the artery wall. computed tomography (CT) scan-a series of cross-sectional X-rays of the brain and head; also called computerized axial tomography or CAT scan. Coumadin®-a commonly used anticoagulant, also known as warfarin. cytokines-small, hormone-like proteins released by leukocytes, endothelial cells, and other cells to promote an inflammatory immune response to an injury. cytotoxic edema-a state of cell compromise involving influx of fluids and toxic chemicals into a cell causing subsequent swelling of the cell. detachable coil-a platinum coil that is inserted into an artery in the thigh and strung through the arteries to the site of an aneurysm. The coil is released into the aneurysm creating an immune response from the body. The body produces a blood clot inside the aneurysm, strengthening the artery walls and reducing the risk of rupture. duplex Doppler ultrasound-a diagnostic imaging technique in which an image of an artery can be formed by bouncing sound waves off the moving blood in the artery and measuring the frequency changes of the echoes. dysarthria-a disorder characterized by slurred speech due to weakness or incoordination of the muscles involved in speaking. dysphagia-trouble swallowing. edema-the swelling of a cell that results from the influx of large amounts of water or fluid into the cell. embolic stroke-a stroke caused by an embolus. embolus-a free-roaming clot that usually forms in the heart. endothelial wall-a flat layer of cells that make up the innermost lining of a blood vessel. excitatory amino acids-a subset of neurotransmitters; proteins released by one neuron into the space between two neurons to promote an excitatory state in the other neuron. extracranial/intracranial (EC/IC) bypass-a type of surgery that restores blood flow to a blood-deprived area of brain tissue by rerouting a healthy artery in the scalp to the area of brain tissue affected by a blocked artery. functional magnetic resonance imaging (fMRI)-a type of imaging that measures increases in blood flow within the brain. glia-also called neuroglia; supportive cells of the nervous system that make up the blood-brain barrier, provide nutrients and oxygen to the vital neurons, and protect the neurons from infection, toxicity, and trauma. Some examples of glia are oligodendroglia, astrocytes, and microglia. glutamate-also known as glutamic acid, an amino acid that acts as an excitatory neurotransmitter in the brain. hemiparesis-weakness on one side of the body. hemiplegia-complete paralysis on one side of the body. hemorrhagic stroke-sudden bleeding into or around the brain. heparin-a type of anticoagulant. high-density lipoprotein (HDL)-also known as the good cholesterol; a compound consisting of a lipid and a protein that carries a small percentage of the total cholesterol in the blood and deposits it in the liver. homeostasis-a state of equilibrium or balance among various fluids and chemicals in a cell, in tissues, or in the body as a whole. hypertension (high blood pressure)-characterized by persistently high arterial blood pressure defined as a measurement greater than or equal to 140 mm/Hg systolic pressure over 90 mm/Hg diastolic pressure. hypoxia-a state of decreased oxygen delivery to a cell so that the oxygen falls below normal levels; see anoxia. incidence-the extent or frequency of an occurrence; the number of specific new events in a given period of time. infarct-an area of tissue that is dead or dying because of a loss of blood supply. infarction-a sudden loss of blood supply to tissue, causing the formation of an infarct. interleukins-a group of cytokine-related proteins secreted by leukocytes and involved in the inflammatory immune response of the ischemic cascade. intracerebral hemorrhage-occurs when a vessel within the brain leaks blood into the brain. ischemia-a loss of blood flow to tissue, caused by an obstruction of the blood vessel, usually in the form of plaque stenosis or a blood clot. ischemic cascade-a series of events lasting for several hours to several days following initial ischemia that results in extensive cell death and tissue damage beyond the area of tissue originally affected by the initial lack of blood flow. ischemic penumbra-areas of damaged, but still living, brain cells arranged in a patchwork pattern around areas of dead brain cells. ischemic stroke-ischemia in the tissues of the brain. lacunar infarction-occlusion of a small artery in the brain resulting in a small area of dead brain tissue, called a lacunar infarct; often caused by stenosis of the small arteries, called small vessel disease. large vessel disease-stenosis in large arteries of the cerebrovascular system. leukocytes-blood proteins involved in the inflammatory immune response of the ischemic cascade. lipoprotein-small globules of cholesterol covered by a layer of protein; produced by the liver. low-density lipoprotein (LDL)-also known as the bad cholesterol; a compound consisting of a lipid and a protein that carries the majority of the total cholesterol in the blood and deposits the excess along the inside of arterial walls. magnetic resonance angiography (MRA)-an imaging technique involving injection of a contrast dye into a blood vessel and using magnetic resonance techniques to create an image of the flowing blood through the vessel; often used to detect stenosis of the brain arteries inside the skull. magnetic resonance imaging (MRI) scan-a type of imaging involving the use of magnetic fields to detect subtle changes in the water content of tissues. mitochondria-the energy producing organelles of the cell. mitral annular calcification-a disease of the mitral valve of the heart. mitral valve stenosis-a disease of the mitral heart valve involving the buildup of plaque-like material on and around the valve. necrosis-a form of cell death resulting from anoxia, trauma, or any other form of irreversible damage to the cell; involves the release of toxic cellular material into the intercellular space, poisoning surrounding cells. neuron-the main functional cell of the brain and nervous system, consisting of a cell body, an axon, and dendrites. neuroprotective agents-medications that protect the brain from secondary injury caused by stroke. oxygen-free radicals-toxic chemicals released during the process of cellular respiration and released in excessive amounts during necrosis of a cell; involved in secondary cell death associated with the ischemic cascade. plaque-fatty cholesterol deposits found along the inside of artery walls that lead to atherosclerosis and stenosis of the arteries. plasticity-the ability to be formed or molded; in reference to the brain, the ability to adapt to deficits and injury. platelets-structures found in blood that are known primarily for their role in blood coagulation. prevalence-the number of cases of a disease in a population at any given point in time. recombinant tissue plasminogen activator (rt-PA)-a genetically engineered form of t-PA, a thrombolytic, anti-clotting substance made naturally by the body. small vessel disease-a cerebrovascular disease defined by stenosis in small arteries of the brain. stenosis-narrowing of an artery due to the buildup of plaque on the inside wall of the artery. stroke belt-an area of the southeastern United States with the highest stroke mortality rate in the country. stroke buckle-three southeastern states, North Carolina, South Carolina, and Georgia, that have an extremely high stroke mortality rate. subarachnoid hemorrhage-bleeding within the meninges, or outer membranes, of the brain into the clear fluid that surrounds the brain. thrombolytics-drugs used to treat an ongoing, acute ischemic stroke by dissolving the blood clot causing the stroke and thereby restoring blood flow through the artery. thrombosis-the formation of a blood clot in one of the cerebral arteries of the head or neck that stays attached to the artery wall until it grows large enough to block blood flow. thrombotic stroke-a stroke caused by thrombosis. tissue necrosis factors-chemicals released by leukocytes and other cells that cause secondary cell death during the inflammatory immune response associated with the ischemic cascade. total serum cholesterol-a combined measurement of a person's high-density lipoprotein (HDL) and low-density lipoprotein (LDL). t-PA-see recombinant tissue plasminogen activator. transcranial magnetic stimulation (TMS)-a small magnetic current delivered to an area of the brain to promote plasticity and healing. transient ischemic attack (TIA)-a short-lived stroke that lasts from a few minutes up to 24 hours; often called a mini-stroke. vasodilators-medications that increase blood flow to the brain by expanding or dilating blood vessels. vasospasm-a dangerous side effect of subarachnoid hemorrhage in which the blood vessels in the subarachnoid space constrict erratically, cutting off blood flow. vertebral artery-an artery on either side of the neck; see carotid artery. warfarin-a commonly used anticoagulant, also known as Coumadin®. The Ischemic Cascade The brain is the most complex organ in the human body. It contains hundreds of billions of cells that interconnect to form a complex network of communication. The brain has several different types of cells, the most important of which are neurons. The organization of neurons in the brain and the communication that occurs among them lead to thought, memory, cognition, and awareness. Other types of brain cells are generally called glia (from the Greek word meaning "glue"). These supportive cells of the nervous system provide scaffolding and support for the vital neurons, protecting them from infection, toxins, and trauma. Glia make up the blood-brain barrier between blood vessels and the substance of the brain. Stroke is the sudden onset of paralysis caused by injury to brain cells from disruption in blood flow. The injury caused by a blocked blood vessel can occur within several minutes and progress for hours as the result of a chain of chemical reactions that is set off after the start of stroke symptoms. Physicians and researchers often call this chain of chemical reactions that lead to the permanent brain injury of stroke the ischemic cascade. Primary Cell Death In the first stage of the ischemic cascade, blood flow is cut off from a part of the brain (ischemia). This leads to a lack of oxygen (anoxia) and lack of nutrients in the cells of this core area. When the lack of oxygen becomes extreme, the mitochondria, the energy-producing structures within the cell, can no longer produce enough energy to keep the cell functioning. The mitochondria break down, releasing toxic chemicals called oxygen-free radicals into the cytoplasm of the cell. These toxins poison the cell from the inside-out, causing destruction of other cell structures, including the nucleus. The lack of energy in the cell causes the gated channels of the cell membrane that normally maintain homeostasis to open and allow toxic amounts of calcium, sodium, and potassium ions to flow into the cell. At the same time, the injured ischemic cell releases excitatory amino acids, such as glutamate, into the space between neurons, leading to overexcitation and injury to nearby cells. With the loss of homeostasis, water rushes into the cell making it swell (called cytotoxic edema) until the cell membrane bursts under the internal pressure. At this point the nerve cell is essentially permanently injured and for all purposes dead (necrosis and infarction). After a stroke starts, the first cells that are going to die may die within 4 to 5 minutes. The response to the treatment that restores blood flow as late as 2 hours after stroke onset would suggest that, in most cases, the process is not over for at least 2 to 3 hours. After that, with rare exceptions, most of the injury that has occurred is essentially permanent. Secondary Cell Death Due to exposure to excessive amounts of glutamate, nitric oxide, free radicals, and excitatory amino acids released into the intercellular space by necrotic cells, nearby cells have a more difficult time surviving. They are receiving just enough oxygen from cerebral blood flow (CBF) to stay alive. A compromised cell can survive for several hours in a low-energy state. If blood flow is restored within this narrow window of opportunity, at present thought to be about 2 hours, then some of these cells can be salvaged and become functional again. Researchers funded by the NINDS have learned that restoring blood flow to these cells can be achieved by administrating the clot-dissolving thrombolytic agent t-PA within 3 hours of the start of the stroke. Inflammation and the Immune Response While anoxic and necrotic brain cells are doing damage to still viable brain tissue the immune system of the body is injuring the brain through an inflammatory reaction mediated by the vascular system. Damage to the blood vessel at the site of a blood clot or hemorrhage attracts inflammatory blood elements to that site. Among the first blood elements to arrive are leukocytes, white blood cells that are covered with immune system proteins that attach to the blood vessel wall at the site of the injury. After they attach, the leukocytes penetrate the endothelial wall, move through the blood-brain barrier, and invade the substance of the brain causing further injury and brain cell death. Leukocytes called monocytes and macrophages release inflammatory chemicals (cytokines, interleukins, and tissue necrosis factors) at the site of the injury. These chemicals make it harder for the body to naturally dissolve a clot that has caused a stroke by inactivating anti-clotting factors and inhibiting the release of natural tissue plasminogen activator. NINDS researchers are currently working to create interventional therapies that will inhibit the effects of cytokines and other chemicals in the inflammatory process during stroke. These brain cells that survive the loss of blood flow (ischemia) but are not able to function make up the ischemic penumbra. These areas of still-viable brain cells exist in a patchwork pattern within and around the area of dead brain tissue (also called an infarct).
|
||||||||||||||||||||||||||||||
|
What
is Tourette syndrome?
What are the symptoms? What is the course of TS? Can people with TS control their tics? What causes TS? What disorders are associated with TS? How is TS diagnosed? How is TS treated? Is TS inherited? What is the prognosis? What is the best educational setting for children with TS? What research is being done? Where can I get more information? What is Tourette syndrome?Tourette syndrome (TS) is a neurological disorder characterized by repetitive, stereotyped, involuntary movements and vocalizations called tics. The disorder is named for Dr. Georges Gilles de la Tourette, the pioneering French neurologist who in 1885 first described the condition in an 86-year-old French noblewoman. The early symptoms of TS are almost always noticed first in childhood, with the average onset between the ages of 7 and 10 years. TS occurs in people from all ethnic groups; males are affected about three to four times more often than females. It is estimated that 200,000 Americans have the most severe form of TS, and as many as one in 100 exhibit milder and less complex symptoms such as chronic motor or vocal tics or transient tics of childhood. Although TS can be a chronic condition with symptoms lasting a lifetime, most people with the condition experience their worst symptoms in their early teens, with improvement occurring in the late teens and continuing into adulthood. What are the symptoms?Tics are classified as either simple or complex. Simple motor tics are sudden, brief, repetitive movements that involve a limited number of muscle groups. Some of the more common simple tics include eye blinking and other vision irregularities, facial grimacing, shoulder shrugging, and head or shoulder jerking. Simple vocalizations might include repetitive throat-clearing, sniffing, or grunting sounds. Complex tics are distinct, coordinated patterns of movements involving several muscle groups. Complex motor tics might include facial grimacing combined with a head twist and a shoulder shrug. Other complex motor tics may actually appear purposeful, including sniffing or touching objects, hopping, jumping, bending, or twisting. Simple vocal tics may include throat-clearing, sniffing/snorting, grunting, or barking. More complex vocal tics include words or phrases. Perhaps the most dramatic and disabling tics include motor movements that result in self-harm such as punching oneself in the face or vocal tics including coprolalia (uttering swear words) or echolalia (repeating the words or phrases of others). Some tics are preceded by an urge or sensation in the affected muscle group, commonly called a premonitory urge. Some with TS will describe a need to complete a tic in a certain way or a certain number of times in order to relieve the urge or decrease the sensation. Tics are often worse with excitement or anxiety and better during calm, focused activities. Certain physical experiences can trigger or worsen tics, for example tight collars may trigger neck tics, or hearing another person sniff or throat-clear may trigger similar sounds. Tics do not go away during sleep but are often significantly diminished. What is the course of TS?Tics come and go over time, varying in type, frequency, location, and severity. The first symptoms usually occur in the head and neck area and may progress to include muscles of the trunk and extremities. Motor tics generally precede the development of vocal tics and simple tics often precede complex tics. Most patients experience peak tic severity before the mid-teen years with improvement for the majority of patients in the late teen years and early adulthood. Approximately 10 percent of those affected have a progressive or disabling course that lasts into adulthood. Can people with TS control their tics?Although the symptoms of TS are involuntary, some people can sometimes suppress, camouflage, or otherwise manage their tics in an effort to minimize their impact on functioning. However, people with TS often report a substantial buildup in tension when suppressing their tics to the point where they feel that the tic must be expressed. Tics in response to an environmental trigger can appear to be voluntary or purposeful but are not. What causes TS?Although the cause of TS is unknown, current research points to abnormalities in certain brain regions (including the basal ganglia, frontal lobes, and cortex), the circuits that interconnect these regions, and the neurotransmitters (dopamine, serotonin, and norepinephrine) responsible for communication among nerve cells. Given the often complex presentation of TS, the cause of the disorder is likely to be equally complex. What disorders are associated with TS?Many with TS experience additional neurobehavioral problems including inattention; hyperactivity and impulsivity (attention deficit hyperactivity disorder—ADHD) and related problems with reading, writing, and arithmetic; and obsessive-compulsive symptoms such as intrusive thoughts/worries and repetitive behaviors. For example, worries about dirt and germs may be associated with repetitive hand-washing, and concerns about bad things happening may be associated with ritualistic behaviors such as counting, repeating, or ordering and arranging. People with TS have also reported problems with depression or anxiety disorders, as well as other difficulties with living, that may or may not be directly related to TS. Given the range of potential complications, people with TS are best served by receiving medical care that provides a comprehensive treatment plan. How is TS diagnosed?TS is a diagnosis that doctors make after verifying that the patient has had both motor and vocal tics for at least 1 year. The existence of other neurological or psychiatric conditions[1] can also help doctors arrive at a diagnosis. Common tics are not often misdiagnosed by knowledgeable clinicians. But atypical symptoms or atypical presentation (for example, onset of symptoms in adulthood) may require specific specialty expertise for diagnosis. There are no blood or laboratory tests needed for diagnosis, but neuroimaging studies, such as magnetic resonance imaging (MRI), computerized tomography (CT), and electroencephalogram (EEG) scans, or certain blood tests may be used to rule out other conditions that might be confused with TS. It is not uncommon for patients to obtain a formal diagnosis of TS only after symptoms have been present for some time. The reasons for this are many. For families and physicians unfamiliar with TS, mild and even moderate tic symptoms may be considered inconsequential, part of a developmental phase, or the result of another condition. For example, parents may think that eye blinking is related to vision problems or that sniffing is related to seasonal allergies. Many patients are self-diagnosed after they, their parents, other relatives, or friends read or hear about TS from others. [1] These include childhood-onset involuntary movement disorders such as dystonia, or psychiatric disorders characterized by repetitive behaviors/movements (for example, stereotypic behaviors in autism and compulsive behaviors in obsessive-compulsive disorder — OCD). How is TS treated?Because tic symptoms do not often cause impairment, the majority of people with TS require no medication for tic suppression. However, effective medications are available for those whose symptoms interfere with functioning. Neuroleptics are the most consistently useful medications for tic suppression; a number are available but some are more effective than others (for example, haloperidol and pimozide). Unfortunately, there is no one medication that is helpful to all people with TS, nor does any medication completely eliminate symptoms. In addition, all medications have side effects. Most neuroleptic side effects can be managed by initiating treatment slowly and reducing the dose when side effects occur. The most common side effects of neuroleptics include sedation, weight gain, and cognitive dulling. Neurological side effects such as tremor, dystonic reactions (twisting movements or postures), parkinsonian-like symptoms, and other dyskinetic (involuntary) movements are less common and are readily managed with dose reduction. Discontinuing neuroleptics after long-term use must be done slowly to avoid rebound increases in tics and withdrawal dyskinesias. One form of withdrawal dyskinesia called tardive dyskinesia is a movement disorder distinct from TS that may result from the chronic use of neuroleptics. The risk of this side effect can be reduced by using lower doses of neuroleptics for shorter periods of time. Other medications may also be useful for reducing tic severity, but most have not been as extensively studied or shown to be as consistently useful as neuroleptics. Additional medications with demonstrated efficacy include alpha-adrenergic agonists such as clonidine and guanfacine. These medications are used primarily for hypertension but are also used in the treatment of tics. The most common side effect from these medications that precludes their use is sedation. Effective medications are also available to treat some of the associated neurobehavioral disorders that can occur in patients with TS. Recent research shows that stimulant medications such as methylphenidate and dextroamphetamine can lessen ADHD symptoms in people with TS without causing tics to become more severe. However, the product labeling for stimulants currently contraindicates the use of these drugs in children with tics/TS and those with a family history of tics. Scientists hope that future studies will include a thorough discussion of the risks and benefits of stimulants in those with TS or a family history of TS and will clarify this issue. For obsessive-compulsive symptoms that significantly disrupt daily functioning, the serotonin reuptake inhibitors (clomipramine, fluoxetine, fluvoxamine, paroxetine, and sertraline) have been proven effective in some patients. Psychotherapy may also be helpful. Although psychological problems do not cause TS, such problems may result from TS. Psychotherapy can help the person with TS better cope with the disorder and deal with the secondary social and emotional problems that sometimes occur. More recently, specific behavioral treatments that include awareness training and competing response training, such as voluntarily moving in response to a premonitory urge, have shown effectiveness in small controlled trials. Larger and more definitive NIH-funded studies are underway. Is TS inherited?Evidence from twin and family studies suggests that TS is an inherited disorder. Although early family studies suggested an autosomal dominant mode of inheritance (an autosomal dominant disorder is one in which only one copy of the defective gene, inherited from one parent, is necessary to produce the disorder), more recent studies suggest that the pattern of inheritance is much more complex. Although there may be a few genes with substantial effects, it is also possible that many genes with smaller effects and environmental factors may play a role in the development of TS. Genetic studies also suggest that some forms of ADHD and OCD are genetically related to TS, but there is less evidence for a genetic relationship between TS and other neurobehavioral problems that commonly co-occur with TS. It is important for families to understand that genetic predisposition may not necessarily result in full-blown TS; instead, it may express itself as a milder tic disorder or as obsessive-compulsive behaviors. It is also possible that the gene-carrying offspring will not develop any TS symptoms. The sex of the person also plays an important role in TS gene expression. At-risk males are more likely to have tics and at-risk females are more likely to have obsessive-compulsive symptoms. People
with TS may have genetic risks for other neurobehavioral
disorders such as depression or substance abuse. Genetic
counseling of individuals with TS should include a full review
of all potentially hereditary conditions in the family. What is the prognosis?Although there is no cure for TS, the condition in many individuals improves in the late teens and early 20s. As a result, some may actually become symptom-free or no longer need medication for tic suppression. Although the disorder is generally lifelong and chronic, it is not a degenerative condition. Individuals with TS have a normal life expectancy. TS does not impair intelligence. Although tic symptoms tend to decrease with age, it is possible that neurobehavioral disorders such as depression, panic attacks, mood swings, and antisocial behaviors can persist and cause impairment in adult life. What is the best educational setting for children with TS?Although students with TS often function well in the regular classroom, ADHD, learning disabilities, obsessive-compulsive symptoms, and frequent tics can greatly interfere with academic performance or social adjustment. After a comprehensive assessment, students should be placed in an educational setting that meets their individual needs. Students may require tutoring, smaller or special classes, and in some cases special schools. All students with TS need a tolerant and compassionate setting that both encourages them to work to their full potential and is flexible enough to accommodate their special needs. This setting may include a private study area, exams outside the regular classroom, or even oral exams when the child's symptoms interfere with his or her ability to write. Untimed testing reduces stress for students with TS. What research is being done?Within the Federal government, the leading supporter of research on TS and other neurological disorders is the National Institute of Neurological Disorders and Stroke (NINDS). The NINDS, a part of the National Institutes of Health (NIH), is responsible for supporting and conducting research on the brain and central nervous system. NINDS
sponsors research on TS both in its laboratories at the NIH
and through grants to major medical institutions across the
country. The National Institute of Mental Health, the
Knowledge about TS comes from studies across a number of medical and scientific disciplines, including genetics, neuroimaging, neuropathology, clinical trials (medication and non-medication), epidemiology, neurophysiology, neuroimmunology, and descriptive/diagnostic clinical science. Genetic studies. Currently, NIH-funded investigators are conducting a variety of large-scale genetic studies. Rapid advances in the technology of gene finding will allow for genome-wide screening approaches in TS, and finding a gene or genes for TS would be a major step toward understanding genetic risk factors. In addition, understanding the genetics of TS genes will strengthen clinical diagnosis, improve genetic counseling, lead to the clarification of pathophysiology, and provide clues for more effective therapies. Neuroimaging studies. Within the past 5 years, advances in imaging technology and an increase in trained investigators have led to an increasing use of novel and powerful techniques to identify brain regions, circuitry, and neurochemical factors important in TS and related conditions. Neuropathology. Within the past 5 years, there has been an increase in the number and quality of donated postmortem brains from TS patients available for research purposes. This increase, coupled with advances in neuropathological techniques, has led to initial findings with implications for neuroimaging studies and animal models of TS. Clinical trials. A number of clinical trials in TS have recently been completed or are currently underway. These include studies of stimulant treatment of ADHD in TS and behavioral treatments for reducing tic severity in children and adults. Smaller trials of novel approaches to treatment such as dopamine agonist and GABAergic medications also show promise. Epidemiology and clinical science. Careful epidemiological studies now estimate the prevalence of TS to be substantially higher than previously thought with a wider range of clinical severity. Furthermore, clinical studies are providing new findings regarding TS and co-existing conditions. These include subtyping studies of TS and OCD, an examination of the link between ADHD and learning problems in children with TS, a new appreciation of sensory tics, and the role of co-existing disorders in rage attacks. One of the most important and controversial areas of TS science involves the relationship between TS and autoimmune brain injury associated with group A beta-hemolytic streptococcal infections or other infectious processes. There are a number of epidemiological and clinical investigations currently underway in this intriguing area. Where can I get more information? For more information on neurological disorders or research programs
funded by the National Institute of Neurological Disorders and
Stroke, contact the Institute's Brain Resources and Information
Network (BRAIN) at: BRAIN Information also is available from the following organizations:
NIH Publication No. 05-2163
|
|
Download
the pdf file: |
Actress's
death puts focus on syndrome -
TRAUMATIC BRAIN INJURY FACTS
SOURCE:
Centers for Disease Control and Prevention The sudden, startling and strange death of actress Natasha Richardson after a seemingly minor fall on a ski slope appears to be an example of “talk-and-die syndrome,” when a traumatic brain injury's initial symptoms are subtle and unnoticed until the patient's condition rapidly deteriorates. “Talk-and-die is rare, but it's a well-known phenomenon,” said Dr. Lawrence Marshall, chief of neurosurgery at the University of California San Diego Medical Center in Hillcrest and an internationally renowned researcher of traumatic head injury. An autopsy conducted yesterday by the New York City Medical Examiner's Office found that Richardson died Wednesday of a brain hemorrhage or clot caused by “blunt impact” to her head. The official cause of death was an epidural hematoma, a swelling of blood between the skull and the dura, which is a thick membrane enveloping the brain. The death was ruled an accident. While the human skull provides a remarkably hard protective case around the brain, it also poses its own dangers. In a fall where the victim's head suddenly and forcefully strikes a surface, the brain – which has the consistency of cooked oatmeal – can be violently sloshed against the sides of the skull, resulting in torn blood vessels and swelling. “Blood starts to accumulate inside the skull, pushing the brain out,” said Mark McDonough, a director at the San Diego Brain Injury Foundation. “But it can only push out so far against the rigid skull. After a while, the skull is basically pushing back. The more pressure builds, the more the skull pushes back, pressing downward on portions of the brain that are critical for basic functions like breathing and heartbeat.” In relatively little time, vital brain regions can be squeezed to failure. In talk-and-die syndrome, experts say, this catastrophic bleeding and swelling are not initially obvious. There are none of the immediate tell-tale indications of brain injury, from mild signs such as headache, nausea, dizziness, blurred vision and confusion to more serious symptoms such as slurred speech, weakness or numbness in limbs, loss of coordination, convulsions or seizures. “That's part of the enigma of brain injury,” said McDonough, an Encinitas clinical psychologist who conducts neuropsychological assessments. “A patient can present relatively well, looking good and coherent, with normal imaging, but the problem is there, escalating.” Richardson, 45, fell while taking a beginning ski lesson Monday afternoon at Mont Tremblant ski resort in Quebec, Canada, about 80 miles northwest of Montreal. Resort officials and observers say the fall appeared to be minor, with Richardson (who was not wearing a helmet) joking about the incident. Nonetheless, her instructor and a ski patrolman escorted her off the slope, and an ambulance was summoned. According to reports, Richardson insisted that she felt fine, and the ambulance was sent away without paramedics examining the actress. Richardson returned to her hotel where, an hour or so later, she complained of a crushing headache and nausea. A second ambulance was called, and it took her to a small hospital in nearby Sainte-Agathe-des-Monts, where she was stabilized. A third ambulance then transported Richardson, who reportedly had lapsed into unconsciousness, to Hopital du Sacre-Coeur near Montreal. Citing Quebec privacy laws, doctors have declined to say what tests (such as a CT scan or MRI) or treatments, if any, Richardson received in Montreal. Marshall noted that no invasive procedures appear to have been done there to relieve dangerously high pressure inside her brain and skull. “Blood clots are the overwhelming problem in talk-and-die cases, but it seems they didn't see anything to operate on,” Marshall said. Twenty-four hours after her fall, Richardson was moved again, this time flown from Montreal on Tuesday afternoon to Lenox Hill Hospital in New York City, near the Manhattan apartment she shared with her husband, actor Liam Neeson. While questions remain about the circumstances surrounding Richardson's accident and death, medical experts say two points are indisputable: First, the actress should have been wearing a helmet. Past studies have shown wearing a helmet while participating in sports such as skiing, snowboarding, bicycling and skateboarding decreases the likelihood of head injury by 40 percent to 60 percent. According to the National Ski Areas Association, an average of 39 skiers and snowboarders are killed annually in the United States, most from head injuries. Helmet use is widely advised at ski resorts but rarely mandatory. Resort operators say they do not want to police customers. Nonetheless, the association estimates that 43 percent of American skiers and snowboarders wore helmets during the 2007-08 season, up from 25 percent five years earlier. Second, time and appropriate care are critical in effectively treating brain injuries. Any symptoms or suspicions of brain trauma should prompt immediate medical evaluation and treatment. “Speed and expertise are everything,” Marshall said. “The earlier the treatment, the better the result. And even in patients who seem OK in the first few hours, experts can often detect something wrong.” The Associated Press contributed to this report. Scott LaFee: (619) 293-1259; Article
used with permission from |
Traumatic Brain Injury
Download
the pdf file: Sandplay
Therapy With Traumatic Brain Injured Adults: |
maintains responsibility for the program. |
|
Learning
Objectives
|
Resources: Brain
Model Tutorial
|
We do adhere to the American Psychological Association's Ethical Principles of Psychologists. Our courses are carefully screened by the Planning Committee to adhere to APA standards. We also require authors who compose Internet courses specifically for us follow APA ethical standards. Many of our courses contain case material, and may use the methods of qualitative research and analysis, in-depth interviews and ethnographic studies. The psychotherapeutic techniques depicted may include play therapy, sandplay therapy, dream analysis, drawing analysis, client and therapist self-report, etc. The materials presented may be considered non-traditional and may be controversial, and may not have widespread endorsement within the profession. www.psychceu.com maintains responsibility for the program and its content. |
All material included in this course is either in the public domain, or used with express permission. |
![]()
|
Thank you! |
To
take the post-test, please return to
http://www.psychceu.com/materialsandtests/login.asp
888-777-3773
|
|
|
|